
Abstract
Introduction
C
PDT is a sequential process that is initiated by the accumulation of the chemotherapeutic agents also known as photosensitizers (PSs) in target cells. Once inside the cells, these chemotherapeutic agents need to be activated, and the cells are irradiated with light at a wavelength specific to the PS. Activated PSs interact with molecular oxygen to generate reactive species (ROS). 7 –9 ROS generated in different subcellular locations promote the generation and stimulation of various intermediates and events that are important factors or checking points in cell death. Therefore, the possibility of regulating tumor cell death becomes an important aspect in monitoring the efficiency of treatment. 10,11
In PDT research, new PSs are constantly being developed in order to improve the efficiency of therapy. Phthalocyanines are a second generation family of PSs with interesting therapeutic characteristics including the following: minimal dark toxicity and healthy cell localization, high neoplastic selectivity, light absorption at a wavelength in the near infrared region, long life and high quantum yield of triplet state, and improved selective neoplastic destruction. 12 These PSs contain a central metal atom that influences the lifetime and the quantum yield of the triplet state of PSs. 13,14 This study investigated the cell death events in a breast cancer cell line (MCF-7) induced by a mixed isomer of sulfonated Phthalocyanine that contains a central Zinc atom (ZnPcSmix).
Materials and Methods
Cell culture
Breast cancer cells (MCF-7 cell line, ATCC® HTB-22), epithelial cells derived from the tissue of a metastatic site of the mammary gland, were used and cultured in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal bovine serum (FBS) (Life Technologies, RSA, Gibco 306.00301), 1% penicillin/streptomycin/fungizone (Scientific Group, RSA, PAA Laboratories GmbH, P11-010) in a 85% humidified atmosphere at 37°C and 5% CO2. When confluent, cells were washed twice with Hank's Balanced Salt Solution (HBSS) (Life Technologies, RSA, Invitrogen, 10-543F) and trypsinised using 1 mL/25 cm2 of TrypExpress (Life Technologies, RSA, Gibco, 15090). A final concentration of 5×105 was used to seed cells in 3.3 cm diameter culture dishes and incubated for 4 h to allow the cells to attach.
Photosensitiser
The PS used in this study was ZnPcSmix; a mixed isomer of sulfonated phthalocyanines, synthesised from (OH2) ZnPc and fuming sulphuric acid by Tebello Nyokong at Rhodes University in South Africa. 15 The ZnPcSmix has a peak absorbance at 680 nm. A concentration of 0.5 μM ZnPcSmix was used to study the mode of cell death in PDT. Approximately 50% of cell death was obtained when this concentration of the PS was used. 5
Laser irradiation
A 680 nm diode laser (Oriel Corporation, USA, LREBT00-ROITHI), provided by the National Laser Centre (NLC) of South Africa, was used for cell irradiation. The FieldMate Laser Power Meter was used to measure the laser output power, which was 52 mW and corresponded to a power density of 5.73 mW/cm2. To avoid light interference, cells were irradiated in the dark without the culture dish lid at room temperature for 29 min 5 sec to deliver an energy density of 10 J/cm2. The laser spot size covered the entire area of the culture dish (9.1 cm2) through a fiberoptic set at 8 cm above the cell monolayer. The parameters of the laser used are presented in Table 1.
Cell cultures were divided into four study groups. Group 1 was an untreated sample (control) that contained no ZnPcSmix and was not irradiated, group 2 (10 J/cm2) contained ZnPcSmix but was not irradiated, and group 3 (0.5 μM) was irradiated but contained no ZnPcSmix. Group 4 (PDT) received ZnPcSmix and was irradiated.
Flow cytometry
Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection (Sigma Aldrich, RSA, APO-AF) was used to determine the cell death events after PDT. The assay uses Annexin V-FITC and Propidium iodide (PI) to detect phosphatidylserine sites on the membrane of apoptotic cells, and sites of membrane damage in necrotic cells, respectively. Therefore, this assay measured two parameters, and gave an overview of the cell population; apoptotic cells (Annexin V-FITC positive, PI negative; or positive for both), necrotic cells (Annexin V-FITC negative, PI positive) and viable cells (Annexin V-FITC negative, PI negative).
Actinomycin D (Sigma Aldrich, RSA, A9415) is a chemotherapy drug that was used as an apoptotic inducer whereas hydrogen peroxide (Sigma Aldrich, RSA, 7722-84-1) was used as an inducer of necrosis at a concentration of 1 μg/mL and 1 mM, respectively. Untreated, ZnPcSmix treated, Actinomycin D treated (apoptotic control), hydrogen peroxide treated (necrotic control), and PDT treated cells were incubated for 24 h prior to Annexin V-FITC and PI staining. After incubation, cells were trypsinized and washed with HBSS. Cells were resuspended in 1X binding buffer at a concentration of 1×106 cells/mL, and 100 μL of the cell suspension was transferred into flow cytometry tubes. Five microliters of each Annexin V-FITC and PI reagents were added, and the tubes were thoroughly mixed and incubated for 10 min at room temperature in the dark. Within 1 h, flow cytometric analysis was performed on the BD FACS Aria (Whitehead Scientific, RSA, BD Biosciences, 642226). Annexin V-FITC was detected as green fluorescence and PI as red.
Cell death enzyme linked immunosorbent assay (ELISA)
A cell death ELISA kit (Roche, RSA, 11 774 425 001) was used to detect and quantify the DNA histone complexes formed during nuclear fragmentation. After treatment, 20 μL of each cell suspension – positive control (DNA-histones complex), negative control (lysis buffer), background control (ABTS substrate buffer), untreated cells, ZnPcSmix treated cells, irradiated cells, and PDT treated cells – was transferred to a 96 well plate. The positive, negative, and background control samples were all supplied with the kit. Then, 80 μL of immune reagent was added to each well, and the microplate was covered with foil and incubated for 2 h at room temperature in a Labcon shaking incubator (Amersham) set at 300 rpm. The solutions were discarded and the wells were rinsed three times with 150 μL of incubation buffer, and then 100 μL of ABTS solution was added to the wells. The microplate was incubated for 20 min at room temperature on a plate shaker (Heidolph Polymax Orbital, Labotec, RSA, 1040) set at 250 rpm. In the final step, 100 μL of ABTS stop solution was added to the wells and the microplate was read at 405 nm and referenced at 490 nm using a Multilabel Counter (Perkin Elmer, VICTOR3™, 1420).
Gene expression
Gene expression analysis was performed using real-time reverse transcriptase polymerase chain reaction (RT-PCR). Complementary DNA (cDNA) samples were used as the templates for RT-PCR and synthesised from a volume containing 30 ng of RNA of untreated cells, ZnPcSmix treated cells, irradiated cells, and PDT treated cells. The RNA samples were isolated using the RNeasy kit (Whitehead Scientific, RSA, Qiagen, 74104) with QIAshredder homogenizers (Whitehead Scientific, RSA, Qiagen, 79654). The synthesised cDNA was used in real-time PCR to study the expression of a panel of genes involved in death and senescence using the Human Cell Death Pathway Finder™ PCR Array (Whitehead Scientific, RSA, SABiosciences, PAHS-212A) on the Stratagene Mx3000p®. The RT 2 Profiler PCR Array profiles the expression of 84 genes (Table 2); all involved in the mechanisms of cellular death. The normalization, fold change, and statistics were performed by the PCR Array data analysis software. The Student's t test was used to calculate the p values from the replicate 2^ (ΔCt) values or fold change for each gene of the untreated cells (control), ZnPcSmix treated cells, and PDT treated cells.
Updated from SABiosciences, PHAS-212A.
Statistics
A breast cancer cell line was used with passages between 10 and 15. Each set of experiments was repeated four times (n=4), excluding PCR (n=3), and each assay was performed in duplicate, with the results being averaged. Untreated cells were included throughout the course of the study, and all treated samples were compared with these cells by means of one way ANOVA, to determine the statistical difference. Statistical analysis was performed using SigmaPlot version 11.0, and the mean, standard deviation, and standard error were obtained. Statistical significances between untreated MCF-7 cells and treated cells are shown as p<0.05 (*), p<0.01 (**), and p<0.001 (***). Significant differences were considered at the 95th percentile.
Results
Flow cytometry
Annexin V-FITC was used together with PI to identify the types of cell population after ZnPcSmix mediated PDT. One hour after staining, flow cytometric analysis was performed, and the majority of the cells in the untreated control and ZnPcSmix treated samples were viable (89% and 91%, respectively) as they stained negative for both Annexin V-FITC and PI (Table 3). However, when compared with untreated control cells, significant changes in cell population were noted with hydrogen peroxide, Actinomycin D, and PDT treated cells. More than 75% of cells treated with Actinomycin D and PDT were undergoing cell death; they were positive for both stains (late apoptotic, p<0.01), positive for Annexin V-FITC (early apoptotic, p<0.01), and the population of normal or viable cells decreased significantly (p<0.001). Changes in necrotic cell populations (positive for PI) were noted with these two populations; however, they were not significant. The change in necrotic cell populations was significant (p<0.01) in the sample treated with hydrogen peroxide.
Data shown are means plus or minus standard errors [±]. Normal cells were both negative for Annexin V-FITC and Propidium iodide (PI), early apoptotic cells were positive for Annexin V-FITC but negative for PI, late apoptotic cells were positive for both Annexin V-FITC and PI, and necrotic cells stained positive for PI. The lowest percentage of cell death (apoptotic and necrotic) was obtained with untreated and Zinc phthalocyanine (ZnPcSmix) controls. Apoptotic populations significantly increased (∼ 65%) in Actinomycin D and photodynamic therapy (PDT) treated cells. With Hydrogen peroxide treatment, the cell death populations (necrotic) significantly increased and the viable population decreased. Experiments were repeated four times (n=4) and significant differences (p<0.01**) and (p<0.001***) were noted compared with the untreated control cells.
Cell death ELISA
Apoptotic cell death is characterized by nuclear fragmentation and oligonucleosomal DNA degradation. A cell death photometric enzyme immunoassay was performed to determine cytoplasmic histone-associated DNA fragments (mono- and oligonucleosomes) subsequent to PDT in MCF-7 cells. No significant quantity of DNA fragments were detected when cells were treated either with laser alone (10 J/cm2) or ZnPcSmix alone (Fig. 1). However, a significant increase in the quantity of DNA fragments as compared with the untreated control was noted after PDT (p<0.01).
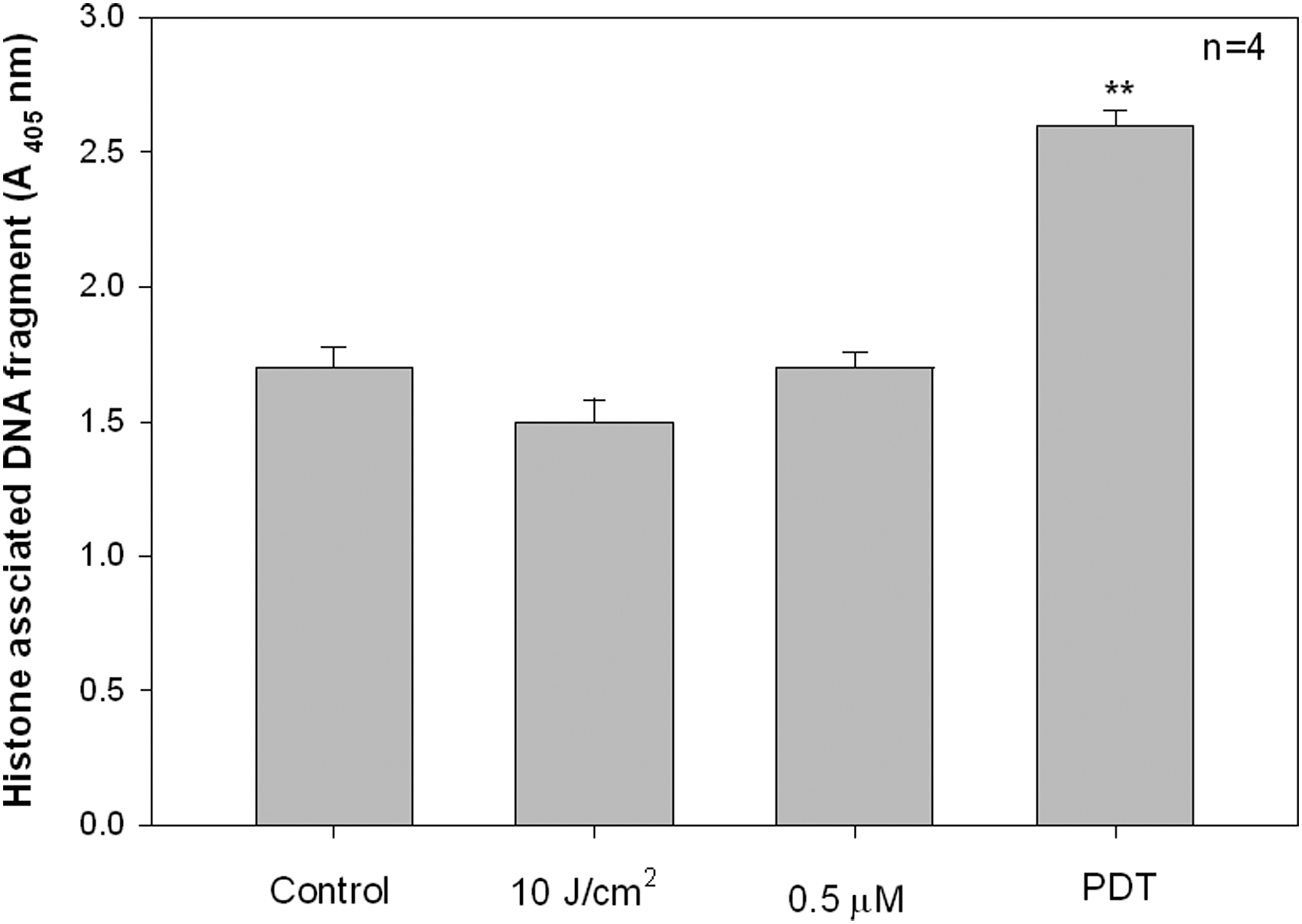
Evaluation of nuclear degradation using the cell death detection enzyme linked immunosorbent assay (ELISA). MCF-7 cells were subjected to various treatment conditions. Untreated (control) and both laser irradiated (10 J/cm2) and zinc phthalocyanine (ZnPcSmix) treated (0.5 μM) cells showed low amounts of DNA fragmentation. After photodynamic therapy (PDT), the amount of DNA fragmentation significantly increased (p<0.01**), compared with the untreated control cells.
Gene expression
MCF-7 cells were treated with 0.5 μM ZnPcSmix alone and incubated for 3 h, before RNA isolation was performed for real-time RT-PCR. Neither the upregulated nor downregulated genes was significantly expressed (not shown). However, after RNA isolation and cDNA synthesis, the gene expression analysis of PDT treated MCF-7 cells showed that B-cell lymphoma 2 (Bcl-2) (p<0.05), DNA fragmentation factor alpha (DFFA)1 (p<0.05) and caspase 2 (CASP2) (p<0.001) were significantly upregulated when compared with the untreated control cells (Fig. 2). None of the downregulated genes were found to be so statistically significant.

Gene expression profiles of photodynamic therapy (PDT) treated MCF-7 cells with 0.5 μM zinc phthalocyanine (ZnPcSmix) and 10 J/cm2 was analysed using the Human Cell Death Pathway Finder Profiler™ PCR Array System. ZnPcSmix mediated PDT induced changes in gene expression and DNA fragmentation factor alpha (DFFA), B-cell lymphoma 2 (Bcl-2) and caspase-2 (casp-2) genes were significantly upregulated as represented in the
Discussion
Cancer death rates have decreased as a result of early detection and treatment, although many of these treatments are still non-cancer specific and they destroy both cancer and normal cells. 16 PDT specifically targets cancer cells and induces cell death via apoptosis, necrosis, autophagy, or a combination of these pathways. 17,18 In normal cells, the phospholipid phosphatidylserines are localized in the cytosolic leaflet of the plasma membrane. 19 Because of damage sustained by cells that result in structural changes, the phospholipid phosphatidylserines relocalize to the outer leaflet. Annexin V binds to phospholipid phosphatidylserines of apoptotic cells, and PI binds to nucleic acid of necrotic cells. 20,21
Cell death mechanisms were studied to determine which cell death mode was induced using ZnPcSmix mediated PDT in breast cancer cells. Flow cytometric analysis revealed that before PDT, most cells remained viable and stained negative for Annexin V-FITC and PI. After PDT or treatment with Actinomycin D, most cells stained positive for these dyes, and apoptotic cells predominated over necrotic and normal cells;>65% of cells stained positive for Annexin V-FITC. This finding was consistent with the work of Chiu and colleagues, 22 which found that apoptotic events were identified after PDT using a silicon phthalocyanine. Apoptosis was also reported to be the induced cell death mode after PDT, and the intrinsic pathway was largely dependent on the caspase activation cascade after the release of cytochrome C and other apoptogenic factors, such as apoptosis-inducing factors (AIF). 23 –25 Apoptosis is induced either through the receptor (extrinsic) or mitochondrial (intrinsic) pathway. 26
Apoptotic cells demonstrate dynamic structural changes, such as chromatin condensation and nucleosomal DNA fragmentation. 27,28 An ELISA was performed to detect and quantify the nucleosomal fragmented DNA bound to antihistone antibodies subsequent to the induction of apoptosis. DNA fragmentation is a well-established marker of apoptosis, and ZnPcSmix mediated PDT led to a significant level of DNA fragmentation in MCF-7 cells. Other studies also reported similar high levels of DNA fragmentation with 88% DNA fragmentation seen 24 h after 5 aminolevulinic acid (ALA) mediated PDT of Jurkat cells. 29 Kessel and Luo observed apoptotic DNA fragmentation 1 h after PDT in murine leukemia cells, using two PSs that localized in lysosomes. 30 During apoptosis, the effector caspase-3 cleaves the inhibitor of caspase activated DNase (ICAD) and releases the caspase activated DNase (CAD), which cleaves DNA between the nucleosomes. DNA fragmentation is a secondary event of apoptosis as a result of the initial action of caspase-3 on ICAD. Real-time RT-PCR was performed to evaluate the gene expression after ZnPcSmix treatment alone and ZnPcSmix mediated PDT on MCF-7 cells. The Human Cell Death Pathway Finder™ PCR Array was used to determine the expression of 84 genes involved in the different cell death pathways. None of the genes in ZnPcSmix treated cells was found to be significantly regulated, but three genes were significantly upregulated after PDT. Bcl-2 (Gene bank ID: NM 000633), casp-2 (Gene bank ID: NM 032982), and DFFA,(Gene bank ID: NM 004401) and all their respective gene products are involved in the apoptotic cell death pathway. Research shows that apoptosis is the mechanism that removes toxins and cell debris, and is often followed by shrinkage and cell fragmentation. 31 It was established that DNA fragmentation factor was required for genomic stability in mammalian cells, and was an important suppressor of cancer. 32
Casp-2 belongs to a family of cysteine proteases that are involved in cell death and inflammatory pathways, 33 and it proteolytically cleaves proteins and associates with other proteins involved in apoptosis. In response to DNA damage, this enzyme also interacts with other proteins to form an activation platform for proteases, known as a PIDDosome, a multiprotein complex consisting of the p53-induced protein with a death domain (PIDD). 34 Casp-2 also induces the release of cytochrome C and other mitochondrial apoptogenic proteins including AIF and secondary mitochondria derived activator caspase (smac) into the cell cytoplasm. This release is enough to activate other mitochondrial apoptosomes in vitro. 35 Caspase is, therefore, a direct effector of the mitochondrial apoptotic pathway.
In response to the high level of mitochondrial apoptogenic proteins such as cytochrome C in the cytosol, Bcl-2 is overexpressed. It was shown that high levels of cytosolic cytochrome C induced the overexpression of Bcl-2. 36 One of the main roles of the Bcl-2 protein is to prevent apoptosis by reducing the release of cytochrome C.
This cell death study showed that Annexin V stained cells were the most abundant after PDT and showed nuclear damage. ZnPcSmix primarily localizes in mitochondria and could have induced mitochondrial damage after irradiation. 5 Such damage possibly caused the release of mitochondrial proteins, including AIF, which mediates nuclear condensation, cytochrome C, and other apoptogenic factors. The release of cytochrome C as a response to the overexpression of casp-2 confirmed the action of apoptotic proteins and the induction of the mitochondrial apoptotic pathway following PDT.
As shown in Figure 3, the proteolytic casp-2 cleaves and activates the Bid into its truncated form (tBid). tBid interacts with proteins, including Bcl-2, on the mitochondrial membrane, and translocates into the mitochondria. 37 –40 This translocation promotes apoptosis by the permeabilization of mitochondrial membranes and release of apoptogenic proteins into the cytosol. Cytochrome C interacts with the apoptotic protease activating factor-1 (Apaf-1) and caspase-9 to form the apoptosome, which further activates cell death effectors including caspase-3, caspase-7, and CAD. 41 –43 Other apoptogenic proteins such as AIF, endonuclease G, and α- and β- DNA fragmentation factors (DFFA and DFFB) lead to nuclear damage and more cell death. 43,44 The induction of cell death and increased level of apoptogenic factors may lead to the overexpression of the Bcl-2 gene encoding for an antiapoptotic protein that inhibits the effects of certain apoptotic stimuli and further cell death induction. 45 –48 However, the actions of caspase independent AIF cannot be prevented by the overexpression of the Bcl-2 gene, and ensure the execution of cell death. 49,50 Apoptosis is a cell death mechanism that removes toxins and DNA fragmentation, and ensures genetic stability and removal of damaged cells.
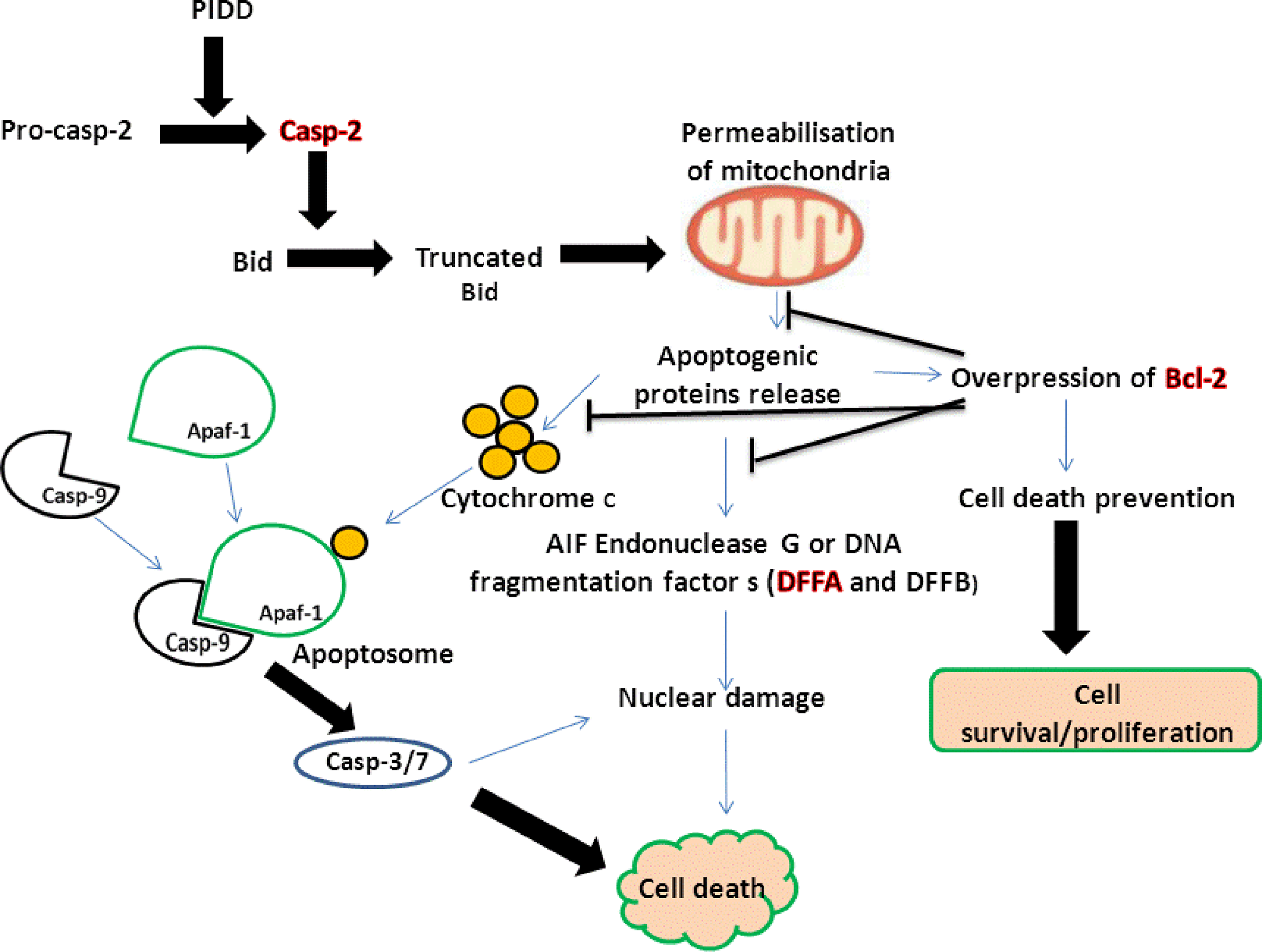
Schematic representation of cell death events. In response to p53-induced death domain associated protein (PIDD) actions, the activated caspase-2 (casp-2) cleaves Bid to form its truncated form (tBid), which induces mitochondrial permeabilization. The release of apoptogenic proteins such as cytochrome C, apoptosis inducing factor (AIF) endonuclease, DNA fragmentation factor alpha and beta (DFFA and DFFB), from the mitochondria lead to further caspase activation, nuclear damage, and cell death. Increased level of apoptogenic proteins can lead to overexpression of the B-cell lymphoma 2 (Bcl-2 gene), which promotes cell survival/proliferation.
Conclusions
ZnPcSmix displayed appreciable photosensitivity and was able to induce cell damage. The study demonstrated that apoptotic cells are the most abundant after PDT and favored by ZnPcSmix, which localized in mitochondria. Disruption of mitochondrial membrane function may be responsible for apoptotic events such as nuclear damage and fragmentation. Furthermore, three genes that are involved in the apoptotic cell death pathway were found to be upregulated. Additionally, casp-2 triggered the release of apoptogenic proteins, which led to more damage. ZnPcSmix mediated PDT effects led to an efficient cell death response in the MCF-7 cell line. The opportunity of controlling the mechanism of cell death has great importance in the clinical context, and the mitochondrial apoptotic pathway can be regulated; therefore, it is a suitable mode of cell death for clinical studies. However, the retention of ZnPcSmix in normal cells and the effects on blood vessels, inflammation, and immune response in situ of neoplasia need to be studied, and if they are found to be limited, future work would be required to determine whether the results obtained in this in vitro study, may be applied in a clinical situation.
Footnotes
Acknowledgments
This work was conducted at the Laser Research Centre at the University of Johannesburg and was supported by the African Laser Centre, National Research Foundation and University of Johannesburg of South Africa. We thank Tebello Nyokong (Department of Chemistry, Rhodes University, South Africa) for the synthesis and supply of the PS. The National Laser Centre of South Africa is acknowledged for the supply and maintenance of lasers.
Author Disclosure Statement
No conflicting financial interests exist.