
Abstract
Introduction
I
The major contributory factors to IR injury are oxidative stress, calcium overload, mitochondrial permeability transition pore (mPTP) opening, and hypercontracture. 3 Myocardial IR injury leads to oxygen-derived free radical production, membrane lipid peroxidation, and impaired post-bypass contractility. 4,5 Oxygen free radicals are cytotoxic molecules generated during reperfusion and/or reoxygenation. Elevated oxygen-derived free radical production initiates and/or promotes apoptotic cascades, leading to cell death and tissue damage. 6 The cell damage induced by reactive oxygen species (ROS) can also initiate a local inflammatory response, which leads to further oxidant stress-mediated tissue damage. 7 Several ROS-producing systems have been identified in many cell types. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, along with other elements of the mitochondrial electron transport chain (ETC), have been reported to be primary sources of ROS production in cardiac tissue. 8,9 Increased ROS induces apoptosis in cardiomyocytes through caspase-3, a cysteine-aspartic acid protease that has been identified as being a key mediator of apoptosis in mammalian cells. 10,11
The existence of lethal IR injury has been inferred in both experimental MI models and in patients by the observation that therapeutic interventions applied solely at the onset of myocardial reperfusion reduced MI size by 40–50%. 3 This observation suggests that lethal IR injury may account for up to 50% of the final MI size. Although improvements in myocardial reperfusion continue to take place, there is still no effective therapeutic strategy for preventing myocardial reperfusion injury.
Photobiomodulation with light in the red to near-infrared (NIR) range (630–1000 nm), generated by using low energy laser or light-emitting diode (LED), augments recovery pathways promoting cellular viability and restoring cellular function following injury. Such photobiomodulation has been observed to increase mitochondrial metabolism, 12 –15 facilitate wound healing, 16 –18 improve recovery from ischemic injury in the heart, 19 and attenuate degeneration in the injured optic nerve. 20 Photobiomodulation promotes angiogenesis in skin, 16 bone, 21 nerve, 22 and skeletal muscle, 23 –26 and modulates apoptotic markers and improves behavioral recovery in a rat model of traumatic brain injury. 27
Zhang et al. have demonstrated that exposure to NIR light at the time of reoxygenation protects neonatal rat cardiomyocytes and HL-1 cardiomyocyte cells from injury, as assessed by lactate dehydrogenase release and MTT assay. 28 Similarly, indices of apoptosis, including caspase-3 activity, annexin binding, and the release of cytochrome c from mitochondria into the cytosol, were decreased after NIR treatment. Treatment with NIR protects cardiomyocytes from hypoxia and reoxygenation injury, in a mechanism dependent upon nitric oxide (NO) generated not only from nitric oxide synthase (NOS), but also from another source of NO, possibly cytochrome c oxidase (CCO). Taken together, these data provide evidence for protection against hypoxia and reoxygenation injury in cardiomyocytes by NIR in a manner that is dependent upon NO derived from NOS and non-NOS sources. 28
Future improvement in delivery of NIR could represent a noninvasive and nonpharmacological therapeutic means to protect against myocardial ischemia and reperfusion injury. The goal of this study was to test the hypothesis that NIR administered at the time of reperfusion decreases the infarct size and increases recovery of mechanical function in rat hearts; measurement of cardiac enzymes and proteins were used as secondary end-points.
Two studies are included in this report. The first is a study of both the safety and efficacy of NIR illumination using the isolated heart model of Langendorff. The major goal was to examine whether NIR itself would pose a safety risk to the functioning of the heart. A secondary goal was to investigate whether NIR would provide some degree of cardioprotection during global ischemia.
The second study was to determine whether NIR would provide cardioprotection in an in vivo model rat model of regional ischemia. Furthermore, we wished to determine if there were any blood markers of cardioprotection that would be translatable to a clinical setting.
Materials and Methods
All experimental procedures and protocols used in this investigation were reviewed and approved by the Animal Care and Use Committee of the Medical College of Wisconsin, Milwaukee, Wisconsin. All conformed to the Guiding Principles in the Care and Use of Animals of the American Physiologic Society and were in accordance with the Guide for the Care and Use of Laboratory Animals. A total of 18 animals were used in the in vitro portion, and 57 animals were used in the in vivo portion of this study. There was a total pre-euthanasia mortality in the in vivo portion of five animals, or 8.8%. Statistical significance was determined by the two-tailed Student's t test.
In groups assigned to receive NIR. radiation, illumination, or therapy, an LED array (Quantum Devices Inc., Barneveld, WI) with a wavelength of 670 nm, 30 nm bandwith, and an irradiance of 66 mW/cm2 (at the LED surface) was used.
In vitro safety and efficacy study
Hearts were assigned to three groups; safety illuminate, efficacy control, and efficacy illuminate (six per group). Isolated rat hearts were perfused retrogradely and instrumented as described previously. 29 The hearts in the safety group were subjected to 10 min exposures of NIR at 670 nm, with the LED array placed on the outside surface of the containment vessel, and with an irradiance of 30 mW/cm2 (measured at the heart surface) for a fluence of 18 J/cm2. Hemodynamic parameters and enzyme leakage [lactate dehydrogenase (LDH)] were measured before illumination, during illumination, and 60 min after illumination.
Hearts in the efficacy groups were subjected to 25 min of global no-flow ischemia and 3 h of reperfusion. The illuminate hearts were exposed at the time of reperfusion in the same manner as the safety group. In addition to hemodynamics and enzyme leakage, infarct size was also measured. The measurements for post-ischemic recovery of left ventricular developed pressure (LVDP) and coronary flow were taken at 60 min reperfusion, and infarct size was determined after 180 min reperfusion. Coronary flow rate was determined by timed collection of the coronary effluent.
In vivo efficacy studies
Male Sprague–Dawley rats (8 weeks old, ∼300 g) were obtained and used for an in vivo anesthetized rat model of cardiac ischemia and reperfusion, in accordance with a previously reported surgical protocol and infarct size (IS) determination method. 30 Briefly, anesthesia was achieved with thiobutabarbital sodium (Inactin, 100 mg/kg, i.p.). Heart rate, blood pressure, and body temperature were continuously monitored. A tracheostomy was performed to facilitate artificial ventilation. The left common carotid artery was cannulated for blood pressure, heart rate, and blood gas measurement. A fifth intercostal space thoracotomy was performed, the pericardium excised, and a ligature placed around the left anterior descending (LAD) artery at approximately the level of the first diagonal branch. Prior to occlusion of the LAD, a pre-injury blood draw of 2 mL was performed for analysis of cardiac troponin I (cTnI), creatine kinase (CK), and LDH via the left common carotid cannula, and a 15-min period was observed for stabilization of the blood pressure. Subsequent occlusion of the LAD was performed by placing the two ends of the ligature through a polypropylene tube, forming a snare, and fixing the snare to the epicardial surface using a hemostat. After 30, 20, 15, or 10 min of occlusion (4–8 per group), the hemostat was removed and the area at risk (AAR) was reperfused for a period of 120 min. In rats assigned to receive NIR therapy, the LED array was positioned 2.5 cm over the AAR beginning 3 min prior to reperfusion and continuing for 7 min after perfusion for a total of 10 min of NIR therapy. Irradiance at the surface of the heart was 43 mW/cm2, for a total fluence of 26 J/cm2. At the completion of the reperfusion period, a 2 mL post-injury blood draw was performed as described. The ligature was again used to occlude the LAD, and the AAR was determined by patent blue negative staining. The heart was then excised, cross-sectioned into five slices, and separated into normal zone and AAR. Slices were incubated in 1% 2,3,5-triphenyltetrazolium chloride to determine infarct size. The heart was incubated overnight in 10% formaldehyde, and the infarcted tissue was dissected from the AAR. Infarct size was expressed as a fraction of the AAR (IS/AAR).
Blood sample processing
Samples for cTnI, CK, and LDH were obtained using “red top” serum collection tubes. Following collection of whole blood, tubes were gently inverted four to five times and allowed to sit upright at room temperature for a minimum of 30 min. Samples were centrifuged at room temperature for 10 min at 3000 rpm. Following centrifugation and inspection for adequate separation of layers, the serum was transferred to 2 mL plastic test tubes and divided to either of two tubes, corresponding to frozen (-20°C) samples (cTnI) or refrigerated (4°C) samples (CK, LDH), respectively. The samples were transferred via courier to an outside facility (Marshfield Labs, Milwaukee, WI) for processing. cTnI was measured using a Siemens Centaur TnI assay.
Results
In vitro safety and efficacy study
Ten minutes of NIR illumination produced no significant differences in the hemodynamic data for the isolated heart safety group (Table 1), whether measured during the illumination or afterwards, or between control and illuminate hearts for the hemodynamic data and infarct size measurements in the isolated heart efficacy groups (Table 2).
Values are mean±SD, n=6/group.
LVDP, left ventricular developed pressure.
Values are mean±SD, n=6/group.
LVDP, left ventricular developed pressure.
In vivo efficacy studies
The study looked at 30, 20, 15, and 10 of LAD occlusion, followed by reperfusion for a period of 120 min with subsequent post-injury blood draw and AAR and IS determination. Serum cTnI levels and IS/AAR were determined for all groups; CK and LDH levels were obtained for the 20 and 30 min groups.
NIR does not affect hemodynamic data
Hemodynamic data for the in vivo study are summarized in Table 3. This includes heart rate, mean arterial blood pressure (MAP), and rate-pressure product (RPP). No significant differences between the NIR groups and their respective control groups were seen, except for the isolated case of RPP for the post-clamp 10 min ischemia groups. There were no significant differences between groups for animal body weight, left ventricular weight, or AAR (data not shown).
Values are mean±SD.
Values for 10 min group taken 10 min post-clamp.
Significant difference (p<0.05) from control.
MAP, mean arterial pressure; RPP, rate-pressure product; NIR, near-infrared.
NIR administered at the time of reperfusion decreases infarct size
NIR treatment at the time of reperfusion produced a significant (p<0.05) decrease in infarct size after 10, 15, 20, and 30 min of LAD occlusion (Fig. 1).
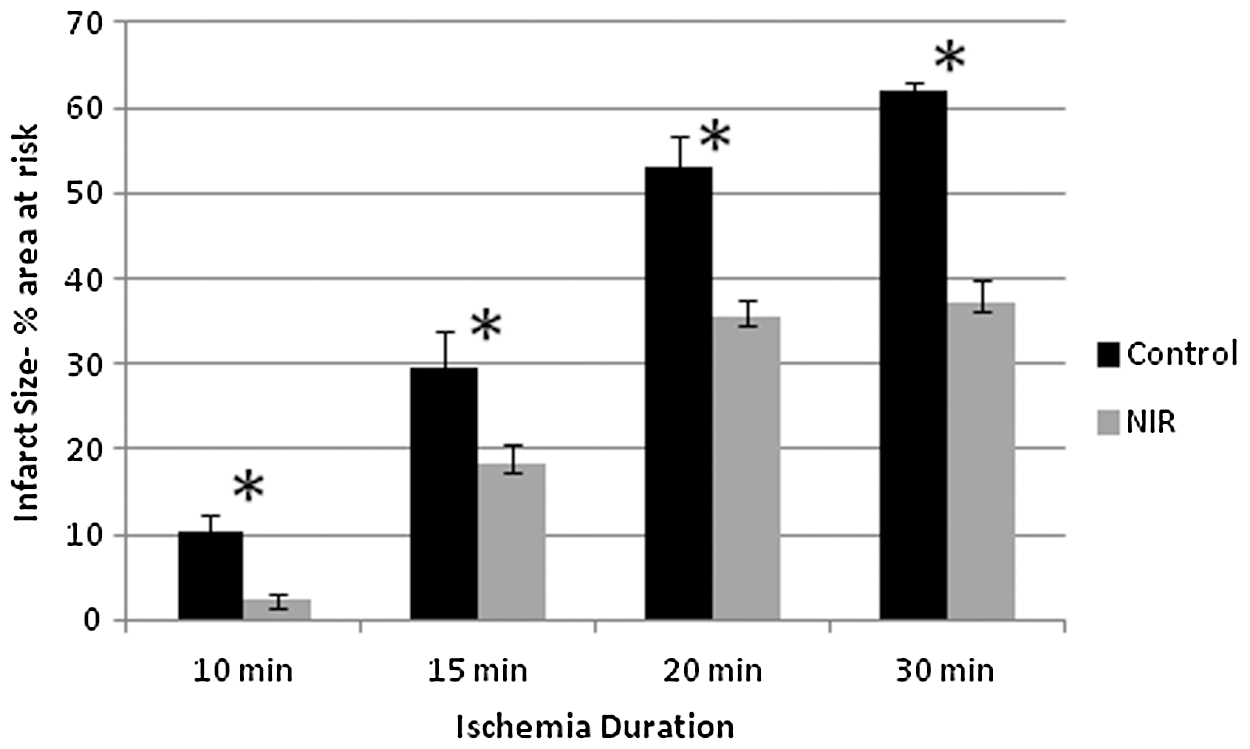
Infarct size measurements as a percentage of area at risk for various periods of ischemia duration, with or without near-infrared (NIR) treatment. Values for infarct size, control versus NIR, are: for 10 min 10.4±4.7 (n=6) versus 2.4±1.6% (n=6), for 15 min 26.8±9.7 (n=7) versus 16.2±6.2% (n=8), for 20 min 54.7±4.4 (n=5) versus 35.4±2.8% (n=7), and for 30 min 62.0±1.1 (n=6) versus 37.1±7.0% (n=7). Values given are mean±standard error. *Indicates significant difference (p<0.05) between NIR and respective control groups.
TnI levels with NIR treatment
The decreases seen in infarct size were not generally matched with a similar decrease in cTnI levels. No significant difference was seen in control versus NIR-treated rat hearts with 10, 20, and 30 min of LAD occlusion (Fig. 2). There was a significant difference between the control and NIR values with 15 min occlusion. cTnI levels showed a high degree of variation between animals within specific groups.
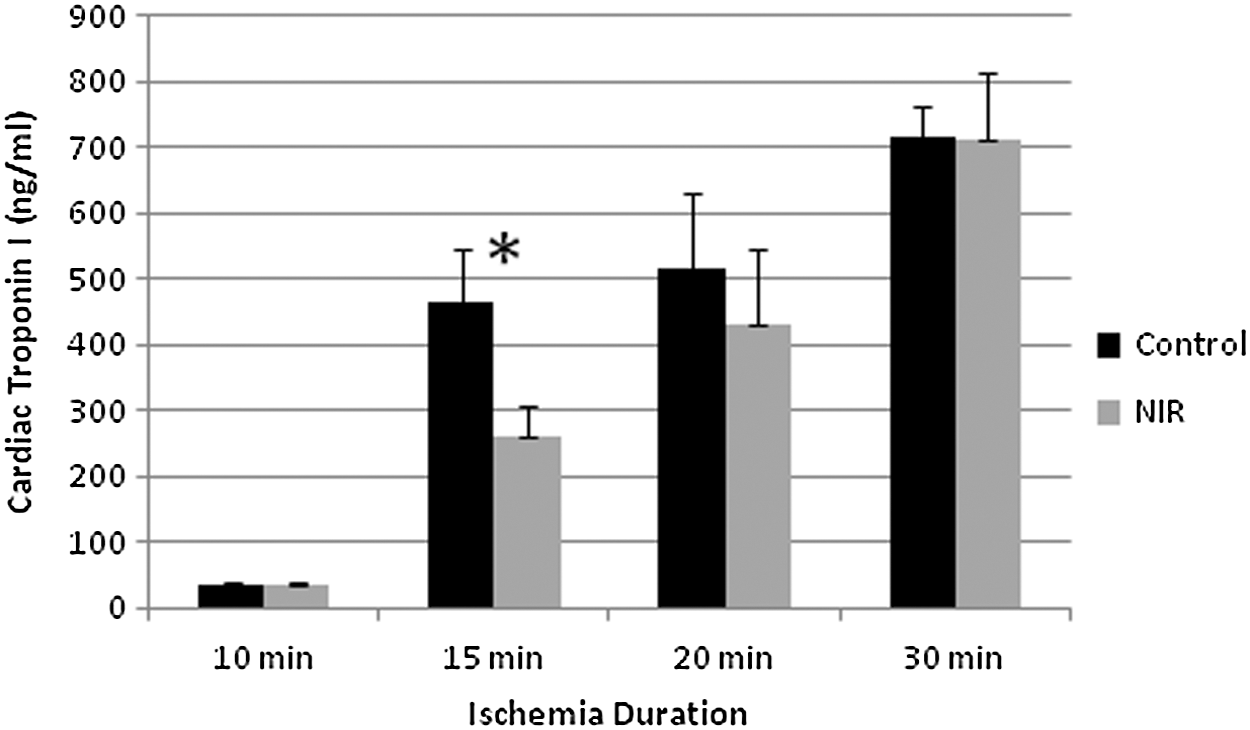
Cardiac troponin I (cTnI) levels (ng/mL) for various periods of ischemia duration, with or without near-infrared (NIR) treatment. Values, control versus NIR, are: for 10 min 34.9±4.8 (n=6) versus 34.5±3.6 (n=6), for 15 min 464±82 (n=7) versus 262±46 (n=8), for 20 min 517±116 (n=4) versus 433±112 (n=5), and for 30 min 715±48 (n=6) versus 714±101 (n=7). Values given are mean±standard error. *Indicates significant difference (p<0.05) between NIR and respective control groups.
No change in CK or LDH levels with NIR treatment
At 20 and 30 min ischemia, CK and LDH levels were also checked as markers of myocardial cell injury. No significant difference (data not shown) was seen in levels of these enzymes to parallel the decrease in infarct size seen with NIR treatment.
Discussion
The study was undertaken for the purpose of exploring the safety of NIR therapy when used for cardioprotection. Furthermore, we wished to determine if there were any biomarkers indicative of cardioprotection that would be useful in a clinical setting. Obviously, typical pre-clinical efficacy measures such as infarct size determination by patent blue staining would be impossible in a phase I clinical trial.
In the isolated heart model, there were no significant differences in hemodynamic values and heart function from pre-illumination to post-illumination, illustrating the basic safety of NIR therapy. However, NIR failed to reduce infarct size after global ischemia.
In the in vivo regional ischemia model, safety was again demonstrated by the hemodynamic data. In contrast to the isolated heart, however, efficacy was seen. Decreases in infarct size related to NIR treatment ranged from 33% to 77% compared with controls. These reductions in infarct size are similar to the cardioprotection afforded by a variety of pharmaceutical and nonpharmaceutical methods of pre- and post-ischemia conditioning, 31 –34 including application of 803 nm laser light in a canine in vivo model. 19 The infarct size reduction seen in the in vivo model but not in the isolated heart model points to the possible involvement of some blood-borne element in NIR-mediated cardioprotection, as the isolated heart is perfused with bicarbonate buffer, or to a fundamental difference between the isolated global ischemia model and the in vivo regional ischemia animal model. This important finding indicates that the clinical setting may be more complex than our pre-clinical models might suggest.
A candidate for the supposed blood-borne element may be iron-nitrosyl hemoglobin or iron-nitrosyl myoglobin as demonstrated by Lohr. 34 Under hypoxic conditions, hemoglobin or myoglobin can reduce nitrite to NO, and then capture this NO in a heme-nitrosyl complex. These complexes are kinetically stable, with slow off-rates. NIR was shown to disassociate the NO from these nitrosyl heme proteins, whereas NIR enhanced the cardioprotective effects of nitrite in an in vivo rabbit model. 34 Furthermore, NIR has been shown to protect isolated cardiomyocytes from hypoxia and reoxygenation injury in an NO-dependent mechanism. 28 This protection, although NO dependent, was only partially dependent on NOS, implying an additional source of NO, possibly by releasing NO from CCO-NO complexes. Although these additional sources of NO may afford susceptibility to NIR-mediated cardioprotection in the isolated cardiomyocyte, they may not be sufficient in the absence of hemoglobin or myoglobin in the blood-free isolated heart.
Consideration of a possible blood-borne element involved in NIR-mediated cardioprotection arising from this study should be qualified with the realization that global and regional ischemia models are different, and are not directly comparable. In regional ischemia, tissue damage is affected by more factors, such as mechanical stress and low-flow collateral circulation, than only lack of oxygen. 35 In addition, global ischemia can be cardioprotective against subsequent regional ischemia, 36 implying the possibility of different damage mechanisms in the two models.
Additional molecular mechanisms can also be postulated to explain NIR-mediated cardioprotection. 37 Many of these other mechanisms also involve NO. Targeting of the cardiolipin–cytochrome c complex by NO can give cardioprotection by inhibiting lipid oxidation. NO can also inhibit the opening of the mPTP upon reperfusion; the opening of the mPTP can initiate cell apoptosis. NO can also reduce ROS generation by targeting the ETC. As the majority of ETC-produced ROS are generated by complexes I and III, inhibition at the level of complex I can reduce ROS, thereby reducing cellular oxidative damage. Conversely, NIR targeting of CCO (complex IV) can afford cardioprotection by stimulating the NOS activity of CCO, 38 or by relieving the NO inhibition of CCO, thereby improving cellular metabolism, adenosine triphosphate (ATP) generation, diminishment of the reduced-cytochrome c pool, and decreased ROS generation. 27,28,39 It appears that both CCO and NO are intimately involved in various ways with the phenomenon of NIR-mediated cardioprotection.
The final major goal of this study was to identify a biomarker of NIR-mediated cardioprotection useful in a clinical trial setting. cTnI has been identified as the most effective translational safety biomarker in this context, 40 and has been proven to be useful in numerous pre-clinical studies. 41 cTnI is considered the biomarker of choice for detection of cardiac cellular injury in clinical use. 42 –45 Although cTnI has a high sensitivity and specificity for myocardial damage, no information regarding the mechanism or the underlying reasons for myocardial injury is given by elevated cTnI levels. 46 The presence of myocardial damage is indicated by elevated cTnI levels, and these increases are associated with an increased risk of adverse events in both the short and long term. 44 –50 In addition, infarct size can be predicted based on cTnI levels. 51 –53 Other biomarkers used in this context include CK and LDH; however, they have been shown to not be as effective as cTnI. 40
In this study, we looked at CK and LDH for the 20 and 30 min occlusion groups, and at cTnI for all groups. CK and LDH showed no significant differences, whereas cTnI showed a significant decrease between control and NIR solely for the 15 min groups, with a lesser, nonsignificant, decrease for the 20 min group. cTnI did show marked increases from pre-ischemia to post-ischemia samples in all groups, with the increases ranging from a 12-fold increase for the 10 min occlusion groups, to an ∼200-fold increase for the 30 min groups. These increases are consistent with previous ischemia and pharmacological toxicity related cTnI increases. 40,41,54 Although we have not unambiguously established the utility of cTnI in measuring cardioprotection by NIR, these results suggest that this may still be attainable, given greater experimental numbers and additional work on decreasing experimental variability.
Conclusions
In sum, we have demonstrated the safety of NIR application in an in vitro rat isolated heart model. In addition, we have demonstrated safety and efficacy when using NIR for cardioprotection in an in vivo rat ischemia model, and have also demonstrated that this cardioprotection is possibly dependent upon some factor present in blood, but not in perfusion buffer. We have not shown the utility of CK, LDH, or cTnI as biomarkers for cardioprotection by NIR, but results show potential for cTnI in this regard. NIR may have therapeutic utility in providing myocardial protection from IR in situations of myocardial ischemia, cardiac surgery, or circulatory arrest.
Footnotes
Acknowledgments
This work was supported by The Bleser Endowed Chair of Neurology (to H.T.W.), The Chad Baumann Neurology Research Endowment (to H.T.W.), and the National Institutes of Health (HL54075 and AI080363 to J.E.B.) We thank Anna Hsu for her expertise in the in vivo rat model.
Author Disclosure Statement
No competing financial interests exist.