
Abstract
Introduction
L
Low-intensity laser irradiation (LILI) is a photobiomodulative therapy that makes use of low intensities of light that is emitted coherently at a specific wavelength in the visible spectrum, which consists of red and near-infrared (IR) light between 600 and 1070 nm, also known as the optical window where effective tissue penetration is maximal. This therapeutic model exposes cells or tissues to visible red light and near-IR light, having either a biostimulatory or a bioinhibitory effect. 7 The mechanism by which LILI induces a photobiological effect is chromophore stimulation at the mitochondrial level, causing various metabolic effects, depending upon the wavelength and fluence used. 8
Previous studies conducted using LILI with wavelengths between 625 and 675 nm and fluences ranging from 1 to 15 J/cm2 all indicated a biostimulatory effect showing an increase in proliferation and viability on various cells; 9 –13 however, studies indicated biochemical inhibition when using higher wavelengths of 800–830 nm and fluences ≥10 J/cm2. 10,14,15
Photobiomodulation relies on specific parameters such as wavelength, fluence, power density, pulse structure, and treatment time when applied to biological tissue. This allows for targeting of specific light-absorbing molecules in specific tissues, operating on the principle of photochemistry, as opposed to photothermogenesis. The light energy absorbed causes singlet state excitation of oxygen molecules, leading to triplet state excitation causing an energy transfer to ground state molecular oxygen (a triplet) to form the reactive species, singlet oxygen. Alternatively superoxide may be formed as a result of electron reduction. LILI operates at an exact wavelength of light, which influences the depth of tissue penetration. Similar to normal cells, cancer cells also contain with intracellular chromophores. Different cellular chromophores are stimulated at different wavelengths. 16,17 Therefore, the prediction can be made that in targeting cancerous cells, the outcome expected can be controlled by the wavelength as well as by the energy output that will lead to either stimulation or inhibition. The exact mechanism behind the stimulation of the light-absorbing molecules producing these two different effects is still being investigated.
CSCs can be identified and isolated using their antigenic markers. 18 Promonin 1 (CD 133) is a pentaspan transmembrane glycoprotein usually found on cell surfaces. 19 It has been used to identify and isolate different SCs and CSCs. 20 Lung CSCs have previously been isolated using the surface marker CD133, 21 and the same antigenic marker was used in this study. Control cell lines used were the CaCo-2 (Caco-2, HTB-37™) cell line, which is a continuous cell of heterogeneous human epithelial colorectal adenocarcinoma cells, which have been found to express CD 133. 20 SKUT-1 (SKUT-1, HTB114™) is a cell line derived from human uterus leiomyosarcoma cells. 22 These cells do not express the surface marker CD 133, as they were used as a somatic cell line and contributed as the negative control cell line. In this study, we aimed to evaluate the biomodulative effects induced by LILI on isolated lung CSCs. It resulted that there were possible detrimental effects associated with LILI when it was used in tissue with underlying malignancy, as LILI provides effective tissue penetration and can produce similar results to the cell layer, as was found in this study.
Materials and Methods
Cell culture
This study used commercially obtained lung cancer cells (A549, ATCC® CCL-185). Rosewell Park Memorial Institute 1640 medium (RPMI) (Sigma, R8758), supplemented with 10% fetal bovine serum (FBS) (Biochrom, S0615), 0.5% penicillin/streptomycin (Sigma, P4333), and 0.5% amphotericin B (Sigma, A2942) was used to culture these cells in. The cells were incubated at 37°C in 5% CO2 and 85% humidity. Cells were grown to confluence before isolation of the CSCs. Upon lung cancer cells reaching confluence, the cells were washed twice with Hank's balanced salt solution (HBSS) (Sigma, H9269) and detached from the culture dish using 1 mL/25 cm2 of TryplExpress (Gibco, 12604). 23 For experimental purposes, isolated lung CSCs were cultured in complete medium using only 5% FBS. Cells were seeded at a final concentration of 2 × 104 cells in 3 mL complete medium, and cultured in a 3.4 cm2 diameter culture dish followed by incubation for 24 h to allow cells to attach.
Control cell lines were cultured in Dulbecco's Modified Eagle's Media (DMEM) (Sigma, D5796) supplemented with 10% FBS, 2 mM L-Glutamine (Sigma, G7513), 1% penicillin/streptomycin (Sigma, P4333), and 1% amphotericin B (Sigma, A2942) for CaCo2 (ATCC®, HTB-37™) which was used as the positive control, 2 and DMEM (Sigma, D5796) supplemented with 10% FBS (Biochrom, S0615), 1% penicillin/ streptomycin (Sigma, P4333), and 1% amphotericin B (Sigma, A2942) for SKUT-1 (ATCC®, HTB114™) as the negative control. 24
Isolation of lung CSCs
Lung CSCs were isolated using the magnetic bead isolation kit (Miltenyi Biotec, QuadroMACS™ separation unit 130-091-051), where they were magnetically labelled with microbead conjugated antibodies directed at the antigenic surface marker of interest. Lung CSCs were enriched using the CD133 MicroBead Kit (Miltenyi Biotec, CD133 MicroBead Kit, human 130-050-801) designed for the positive selection of cells expressing human CD133 antigen.
A single cell suspension was prepared, where the cell number was determined. The cells were spun down and resuspended in 80 μL buffer per 107 total cells, and 20 μL MicroBeads per 107 total cells were added and incubated for 15 min at 2°–8°C. Cells were washed, centrifuged, and resuspended in 500 μL buffer per 108 cells. The separation column was prepared by running buffer through and discarding the effluent. The cell suspension was applied to the prepared column, where the unlabelled cells were collected and discarded and the positively selected cells in the column were flushed out into a suitable collection tube using buffer and a plunger. 25
Immunofluorescence
Fluorescent labelling of surface antigens with CD 133 (Prominin-1), which is a monoclonal antibody hosted in mouse and reacting in humans (USBio, 030034) on isolated cells, was used to confirm whether the lung CSCs that were isolated were of CSC origin. 26 Isolated cells along with control cell lines CaCo2 and SKUT-1 were cultured on heat- sterilized cover slips placed in Petri dishes (3.5 cm diameter), at a concentration of 2 × 105 cells in complete media. Cells were incubated overnight to attach to the cover slip. Cultures were removed from the incubator and washed twice with ice-cold phosphate-buffered saline (PBS)/bovine serum albumin (BSA)/azide buffer (PBS, Sigma, P4417), 0.1% w/v BSA (Sigma, A2153), and 0.01% w/v azide (Sigma, S8032) on ice, and then incubated with 10% serum (Normal Goat Serum, abcam, ab7481) for 30 min on ice as a blocking step, and then washed twice again. Cells were then incubated with 100 μL primary antibody rabbit anti-human CD 133+ (Abnova, PAB12663) in working buffer (2% serum in PBS BSA/azide buffer) for 30 min on ice. Cells were then rinsed three times with PBS BSA/azide buffer and incubated with 100 μL of the secondary fluorescent antibody (abcam, ab6717) in working buffer for 30 min on ice, protected from light. Cells were rinsed three times as before and fixed in 4% parafomaldehyde for 10 min. After fixation, cells were rinsed once briefly with PBS, and then once with tap water before being stained with 4′-6-diamidino-2-phenylindole (DAPI) (Sigma, D9564) and mounted on glass slides using 0.1 M propylgallate (Sigma, 02370). Slides were viewed using a fluorescent microscope (Carl Zeiss, Axio Observer Z1). 14
Laser irradiation
After 24 h of incubation, the culture medium was removed and the cells were washed with HBSS. Fresh medium was added, and cells were irradiated using a 636 nm diode laser (LTIAO00-PLT20-636nm) provided by the National Laser Center (NLC) of South Africa. The power output was measured using the FieldMate Laser Power Meter (Detector S/N: 125J07R) and the value was used to calculate the exposure time. Cells were irradiated with energy densities of 5, 10, and 20 J/cm2. Cells were irradiated from above, at room temperature with the culture dish lid off. Irradiation was performed in the dark, omitting nuisance variables that would interfere with the laser effect, such as polychromatic light. Laser parameters are shown in Table 1. Cell cultures were divided into four study groups. Group 1 was an unirradiated control, group 2 was irradiated at 5 J/cm2, group 3 was irradiated at 10 J/cm2, and group 4 was irradiated at 20 J/cm2. Post-irradiation, cells were incubated for 24, 48, or 72 h.
Cell morphology
Morphological changes in the four different study groups of isolated lung CSCs were observed and studied using an inverted light microscope (Wirsam, Olympus CKX41) 24, 48, or 72 h post-irradiation. Morphological pictures were taken with the SC30 Olympus camera.
Adenosine triphosphate (ATP) cell viability assay
The number of metabolically active cells was determined using the CellTiter-Glo® luminescent cell viability assay (Whitehead Scientific, Promega, G7573). This homogeneous method determines the number of viable cells in culture based on ATP quantitation. The assay utilizes the properties of a proprietary thermostable luciferase, which generates a luminescent signal proportional to the amount of ATP present released upon cell lysis. According to the manufacturer's protocol, 50 μL of reconstituted reagent was added to an equal volume of cell suspension. The contents were mixed in a shaker for 2 min to induce cell lysis. The contents were then incubated at room temperature for 10 min to stabilize the luminescent signal. 27 The amount of ATP was quantified, and luminescence was recorded using the Perkin Elmer, VICTOR3™ Multilabel Counter (model 1420) in relative light units (RLUs).
Trypan blue exclusion assay
Trypan blue staining method was used to quantify percentage viable cells. The assay is employed to identify dead cells, as cells that are viable have intact membranes and can effectively exclude the dye, whereas dead cells with compromised membranes become stained. Equal volumes of 0.4% Trypan blue (Invitrogen™, Trypan Blue Stain [0.4%] T10282) and cell suspension were mixed and loaded into a counting chamber, where the number of cells that were viable and dead was counted using the Countess® Automated Cell Counter. Percentage viability was determined by calculating the number of viable cells from the total number of cells.
Alamar blue cell proliferation assay
Alamar blue is a quantitative assay that uses a fluorometric growth indicator based on detection of metabolic activity. It uses a redox indicator, where metabolically active cells will reduce the dye Resazurin, which is blue, to its reduced form Resorufin, which is a fluorescent red. Then, 100 μL of cell suspension was added to a 96 well microtiter plate, and 10 mL AlamarBlue® reagent (Invitrogen™, AlamarBlue® DAL1025) was added. The plate was incubated for 3 h (37°C in 5% CO2). Fluorescence was then measured using the Victor-3 (Perkin-Elmer, Separation Scientific) at Ex/Em 560/590.
Statistical analysis
The isolated lung CSC cell lines were used between passage numbers one and four. Experiments were repeated four times (n = 4). All assays were performed in duplicate, and the average was used. Results were statistically analysed using Sigma Plot Version 8.0, and the mean, standard deviation, and standard error were obtained. A student t test and one way ANOVA were performed to detect differences between the control and experiments and between experimental groups, as well as differences between controls and treated samples from 24 to 72 h. Statistical significances are indicated in the figures as p < 0.05 (*), p < 0.01, (**) and p < 0.001 (***).
Results
Immunofluorescence
Expression of the CD 133 stem cell marker after CSC isolation was determined by indirect immunofluorescence (Fig. 1). Isolated lung CSCs expressed the surface marker CD 133, as well as the positive control cell line CaCo2. No fluorescence was observed from the negative control cell line SKUT-1. DAPI was used to counterstain the nuclei. Positive expression of the CD 133 antigenic surface marker indicated positive isolation of lung CSCs.
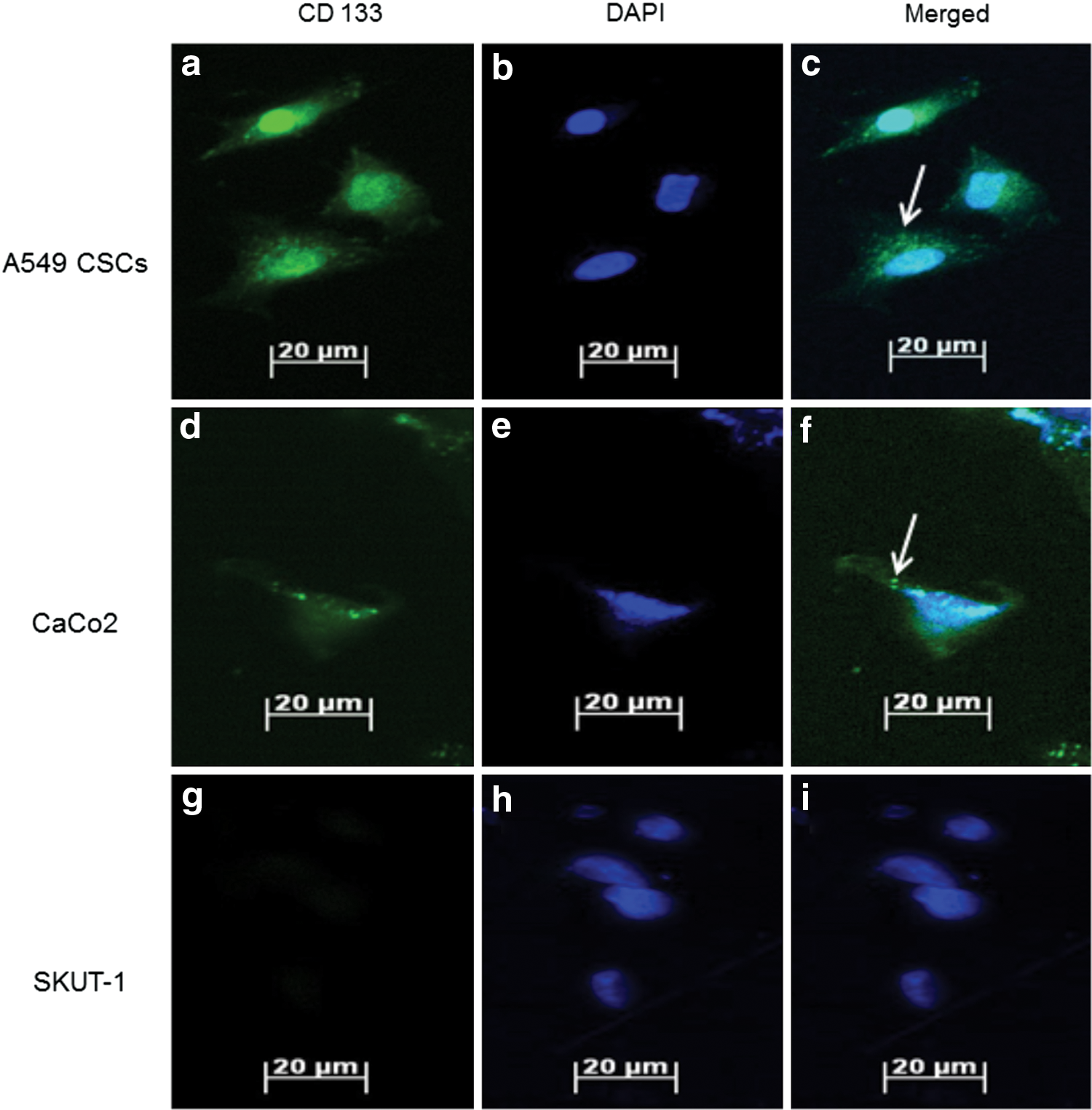
Characterisation of isolated A549 cancer stem cells (CSCs). CD 133 antibody is fluorescein isothiocyanate (FITC) bound, staining the cell surface green (
Cell morphology
Lung CSC morphological changes are demonstrated in Fig. 2. Unirradiated control cells demonstrated no morphological changes even after several time intervals, and maintained their cell shape and size (a, b, and c). Irradiated cells exposed to fluences of 5, 10, and 20 J/cm2 did not present any morphological disparity compared with untreated control cells (d–l). Changes were observed in cell density, which increased over time as cells proliferated. All cells grew as a monolayer with epithelial morphology with strong proliferation and adhesion properties.
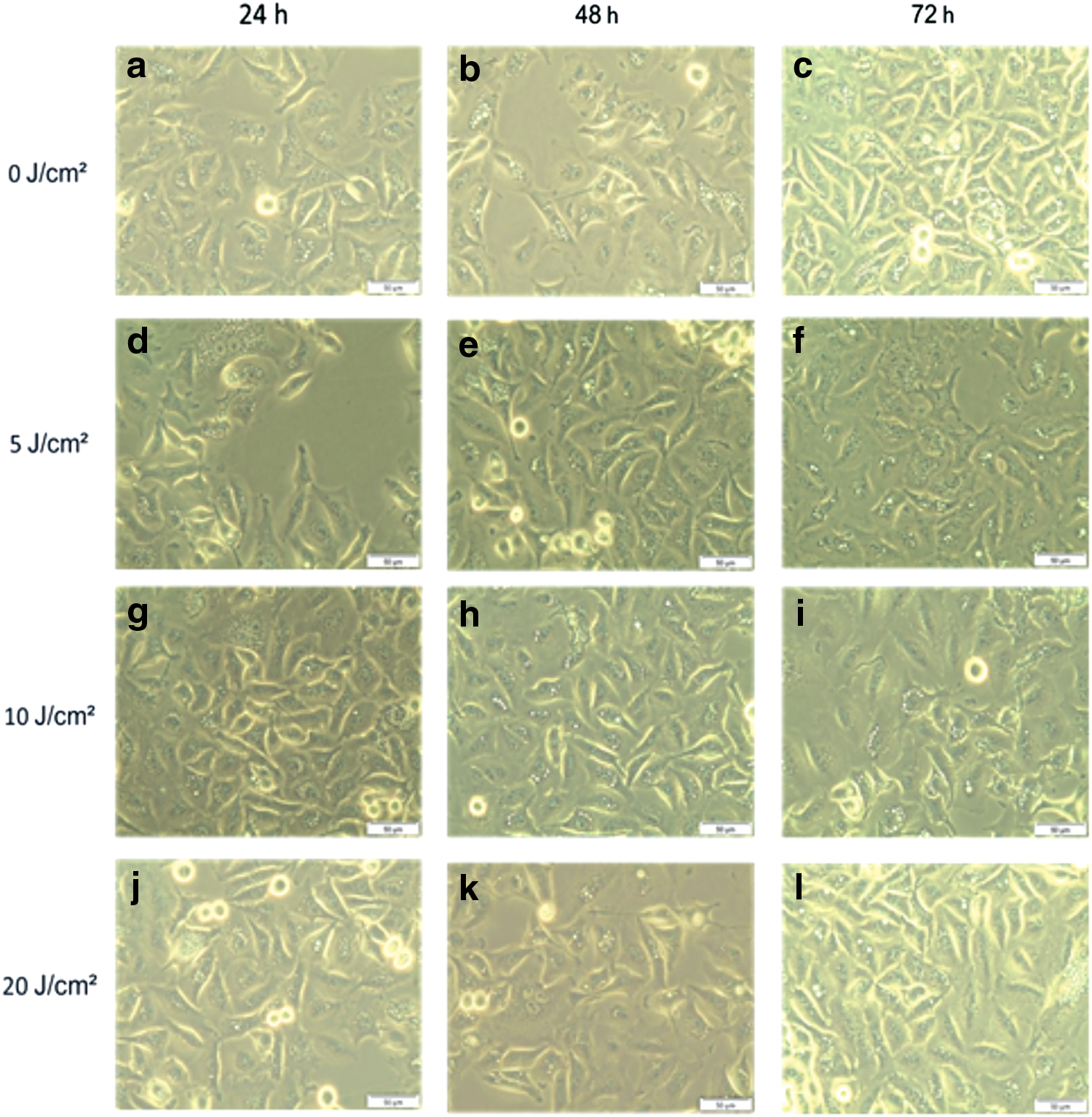
Morphology of A549-isolated lung cancer stem cells (CSCs). Control cells received no irradiation (
ATP cell viability assay
Post-irradiation cellular viability was determined by ATP luminescence (Fig. 3). Significant differences were observed between the control and irradiated cell sample when comparing the viability from 24 to 72 h for 5 and 10 J/cm2 as indicated in Fig. 3. A statistically significant increase in viability was seen when comparing the test samples with each other for 20 J/cm2 at 72 h. This may be indicative of the initial suppression of viability caused by the higher fluence at 24 h, but because of CSC self-renewal, viability increased significantly from 24 to 72 h. Irradiated cells show a stimulatory trend in viability when compared with their unirradiated controls over time, although the differences between the these cells were not statistically significant.
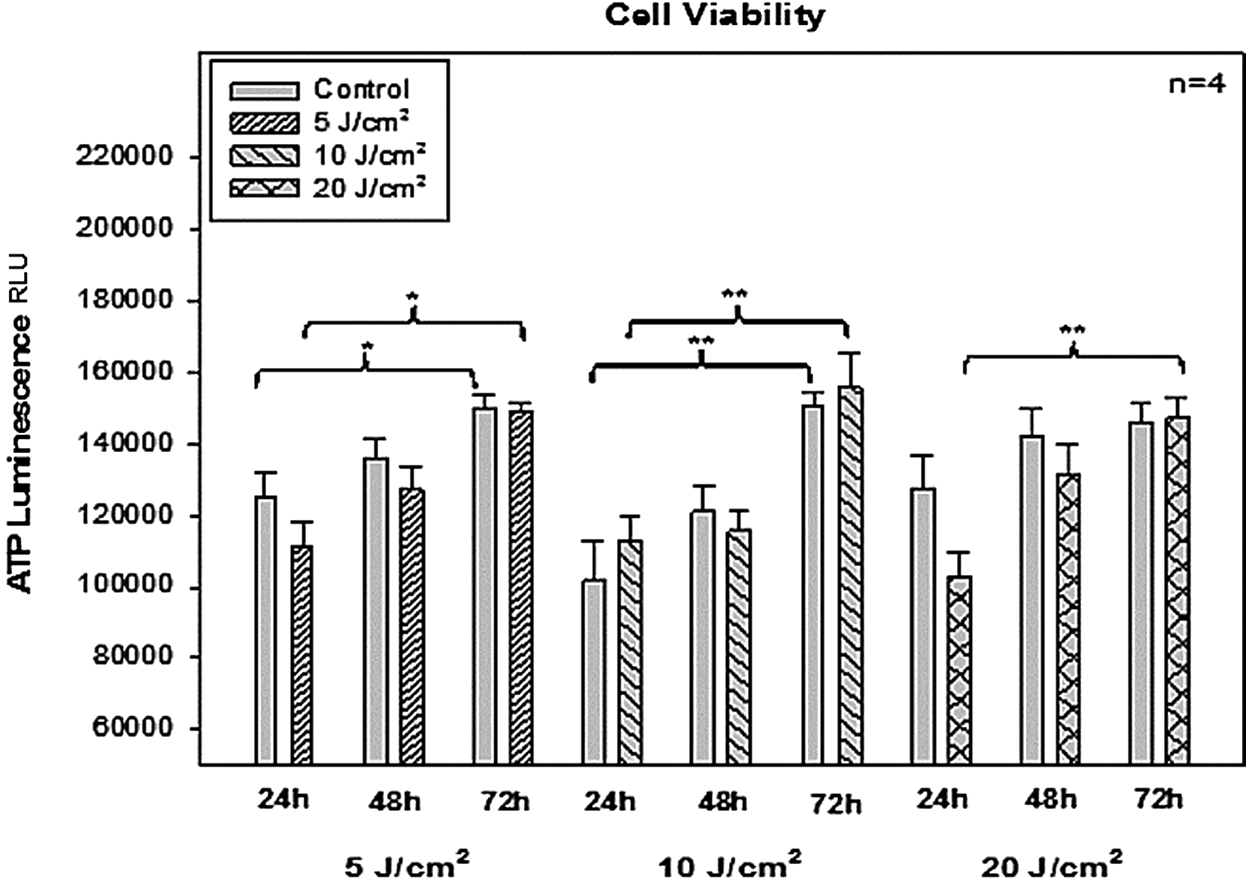
Increase in adenosine triphosphate (ATP) luminescence is seen in all the samples, as time elapses from 24 to 72 h and statistical significances are indicated. No significant change was observed when comparing 5, 10, and 20 J/cm2. All samples were compared with their control groups. Experiments were repeated four times (n = 4).
Trypan blue exclusion assay
Trypan blue exclusion is used to estimate the proportion of viable cells and nonviable cells in a population. The percentage viability of cells is demonstrated in Table 2. When examining Trypan blue values, statistically significant changes were observed when comparing each fluence and its viability over 72 h. At 5 J/cm2, a greater increase in viability was observed in the irradiated group when compared with its control over time from 24 to 72 h, p < 0.001. The opposite is seen when comparing 20 J/cm2, with irradiated cells having a lesser increase in viability.

AlamarBlue™ cell proliferation assay
AlamarBlue™ was used to measure the number of proliferating cells (Fig. 4). Measurement was read as an absorbance value, with an increase in proliferation over several hours observed in all samples. When comparing unirradiated samples to treated samples, a statistically significant difference was observed in all fluences used on the cells during their proliferation time of 24–72 h post-irradiation (p < 0.001).

The Alamar blue assay was used to measure cell proliferation. Increase in proliferation is seen in all the samples as time elapses from 24 to 72 h. Unirradiated cells' proliferation time was compared with the treated cells' proliferation time. Statistical significant differences were expressed as p < 0.001 (***).
Discussion
Because the hypothesis states that CSCs are a contributing factor to tumor initiation, metastasis, and cancer relapse, 28 it raises concern not only when considering current cancer treatments, but also about LILI when used as a therapeutic model. Cancer treatments such as surgery, radiotherapy, and chemotherapy still offer restricted prognostic outcome when it comes to competence, and produce undesirable side effects. Therefore, alternate therapeutic options are essential, and need to be investigated. 23 The focus needs to be turned to CSC-targeted therapy. LILI on the other hand which has been used for its desired effects of increased cell proliferation and viability, 9 can be harmful, bearing in mind CSC characteristics.
Previous studies conducted on lung cancer indicated that CD 133 positive cells displayed a greater ability for self-renewal, tumor initiation, and drug resistance. 29 In previous studies conducted using neuroblastoma (NB) tumor samples expressing CD133, the marker was shown to repress cell differentiation and accelerated cell proliferation, anchorage-independent colony formation, and in vivo tumor formation, 30 all indicative of CSC characteristics.
In this study, we explored the possibility of outcomes using LILI on lung CSCs. Lung CSCs were successfully isolated using the CD 133 cell surface marker. Control cells receiving no irradiation maintained their cell morphology, whereas there was an increase seen in viability and proliferation as a result of normal cell cycle maintenance. These results concur with those of Mvula et al. 13 In this study, when comparing irradiated CSCs to their unirradiated controls at 24, 48, and 72 h, there were no statistical differences seen at specific time intervals when assessing viability and proliferation (Figs. 3 and 4, Table 2). However, when comparing viability and proliferation of controls at 24 h with controls at 72 h, and the same for irradiated samples at 24 h versus irradiated samples at 72 h, a statistically significant increase for both viability and proliferation was seen at 5 and 10 J/cm2.
No significance was seen between controls at 24 h with controls at 72 h at 20 J/cm2 when analyzing ATP viability results (Fig. 3), although statistically significant increases were seen after analyzing Trypan blue viability (Table 2) and Alamar blue proliferation (Fig. 4) in the same samples. Significant increases were seen in viability and proliferation when comparing irradiated samples at 24 and 72 h for 20 J/cm2, showing a significant stimulation in the cell cycle leading to an exponential rate in mitosis. When considering the average increase in viability and proliferation of controls for three periods, viz., 24, 48, and 72 h, and one compares that to the average increase for the irradiated samples, there is also a statistically significant increase in the irradiated samples. There were changes in cell morphology, increased cell viability, and increased cell proliferation. These results can be explained by the following: previous studies suggest that LILI can stimulate cells with a wavelength of 636 nm and a low fluence of 5 J/cm2. 13 One SC characteristic is that they stay quiescent until regeneration of cells stimulates the proliferation via a specific signal, whereas CSCs have the ability to self-renew and proliferate indefinitely disregarding cell signalling pathways that may cause a cell to stop self-renewal or proliferation, 31 allowing the unirradiated control CSCs and irradiated CSCs to proliferate at a similar rate. CSCs are capable of self-renewal opposing cell death, 6 even when exposed to higher fluences of 20 J/cm2, suggesting that irradiation at 5, 10, and 20 J/cm2 with a wavelength of 636 nm has no damaging effect, but rather a proliferatory effect on lung CSCs. This is then in agreement with previous studies also indicating that parameters such as wavelength, fluence, and intensity play an important role in photobiomodulation of cellular metabolism, 10,12 –14 which shows that wavelengths between 600 and 800 nm with fluences of 5–20 J/cm2 can induce biostimulation, whereas wavelengths of 800–1070 nm with fluences of 20–40 J/cm2 may be able to induce bioinhibition.
Conclusions
This study indicates the possible detrimental effect that LILI may have when used as a biostimulatory therapy on the underlying tissue CSCs when considering the proliferation and viability induced using visible wavelengths. Therefore, LILI should be used with caution clinically, bearing in mind the risk of stimulating potential underlying CSCs, as LILI provides optimum penetration depth in tissue, reaching underlining cell layers, which may react similarly to in this study. When considering the use of LILI as a bioinhibitory treatment of human lung cancer, one needs to explore these effects on lung CSCs, using higher wavelengths such as IR, because visible red light initiates CSC proliferation and viability. This study focused on lung CSCs at 636 nm. Other CSCs may respond differently than lung CSCs as it has been indicated in several studies that cancer cell lines differ with respect to their response to photobiomodulation. We propose that additional research should be conducted to establish the effects of LILI on lung as well as additional CSCs, determining the effect of wavelength and fluence.
Footnotes
Acknowledgments
This work was conducted at the Laser Research Centre at the University of Johannesburg and was supported by the National Laser Centre of South Africa.
Author Disclosure Statement
No competing financial interests exist.