
Abstract
Enzyme replacement therapy is an established means of treating lysosomal storage diseases. Infused enzymes are normally targeted to the lysosomes of affected cells by interactions with cell-surface receptors that recognize carbohydrate moieties such as mannose and mannose 6-phosphate on the enzymes. Therefore, we have investigated alternative strategies to deliver the lysosomal enzyme β-glucuronidase in the enzyme-deficient mucopolysaccharidosis type VII mouse model. Here we summarize our recent efforts to use nontraditional ways to deliver β-glucuronidase. First, we used a chimeric protein of the insulin-like growth factor II (IGF-II) fused to β-glucuronidase to deliver enzyme via the IGF-II binding site on the bifunctional IGF-II/mannose 6-phosphate receptor. Second, we used the 11-amino-acid human immunodeficiency virus (HIV) Tat domain fused to β-glucuronidase to mediate uptake by absorptive endocytosis. Interaction with heparan sulfate on the cell surface internalizes and delivers the Tat-tagged enzyme to the lysosome via plasma membrane recycling. Third, we created a chimeric β-glucuronidase fused to the Fc portion of human immunoglobulin G (IgG) Fc, which was transported by the neonatal Fc receptor from the maternal circulation across the placenta to sites of storage in fetal tissues. Finally, periodate treatment was used to eliminate interaction with carbohydrate receptors, creating an enzyme with increased plasma half-life, resulting in transport across the blood–brain barrier and clearance of storage in neurons. These strategies for delivering lysosomal enzymes could also be used to target nonlysosomal proteins or enzymes identified for bioremediation of other conditions.
Introduction
The classical example is the means by which the enzyme glucocerebrosidase, which is deficient in Gaucher disease, is delivered to macrophages. 2,3 Delivery of this enzyme is dependent on the binding of terminal mannose residues located on the carbohydrate chains of the enzyme. These residues bind to mannose receptors (MRs) on the cell surface of macrophages and other cells of the reticuloendothelial system. After endocytic internalization, the enzyme is transported by endosomes to the lysosome, resulting in the breakdown of accumulated storage material.
In contrast to Gaucher disease, most other LSDs contain storage in other cell types that lack the MR. Delivery to these cell types employs a different receptor, the insulin-like growth factor II/cation-independent mannose 6-phosphate receptor (IGF-II/MPR). This receptor recognizes mannose 6-phosphate (M6P) moieties present on the carbohydrate chains of lysosomal enzymes synthesized in mammalian cells. 4 Enzymes such as β-glucuronidase (GUS), α-iduronidase, and α-galactosidase, which are deficient in mucopolysaccharidosis VII (MPS VII), MPS I, and Fabry disease, respectively, rely on this delivery system to deliver infused enzyme by the IGF-II/MPR present on the cell surface. 5 In addition, these enzymes usually have carbohydrate chains containing terminal mannose residues, which allow uptake by the MR.
In many cases, lysosomal enzymes used for ERT produce less than ideal results. In some cases, enzymes such as α-glucosidase, which is deficient in Pompe disease, are very poorly phosphorylated when the recombinant enzyme is produced in mammalian cell systems. This condition requires the use of very high doses of enzyme to obtain even modest clearance of storage in these patients. 6 In other cases, rapid clearance of infused enzyme from the circulation by IGF-II/MPR and MR in the liver and spleen lead to inadequate delivery to other tissues. Another problem is the lack of delivery of enzyme across the blood–brain barrier (BBB). This is due to the natural function of the BBB to limit access by unwanted substances and to the lack of both the MR and IGF-II/MPR transport systems at that location.
To improve delivery to resistant sites, we have investigated alternative strategies for delivery of lysosomal enzymes using the lysosomal enzyme GUS in the murine model of β-glucuronidase deficiency. For these studies, we used the enzyme-deficient MPS VII/E540A tg mouse model that we previously made tolerant to human GUS. 7 This tolerance allows multiple infusions of the human enzyme to be used without complications arising due to immunological reaction.
Our initial experiment involved a peptide based targeting system known as GILT, (
A second strategy was the use of the 11-amino-acid human immunodeficiency virus (HIV) Tat protein transduction domain to make a carboxy-terminal fusion protein with GUS (GUS-Tat). 9 We found that GUS-Tat could be taken up in an M6P-independent manner by interaction of the positively charged Tat peptide with cell-surface proteoglycans.
Another objective was to treat the storage that begins during prenatal life in many patients with MPS. It is known that maternal immunoglobulin G (IgG) is transported across the placenta by the neonatal Fc receptor (FcRn), which recognizes the Fc domain on IgG. We hypothesized that combining the Fc-tag to the missing enzyme might deliver it across the placenta to the fetal circulation. To test this hypothesis, we made a fusion protein in which a portion of the IgG Fc domain was fused to the carboxyl terminus of GUS (GUS-Fc). Subsequently, we infused GUS-Fc into pregnant MPS VII mice and found that it could be transported across the placenta and delivered to the fetus. 10
Another aim was to attain correction of lysosomal storage in the brain, which has in the past been limited by the inability of infused enzyme to cross the BBB. Earlier studies indicated that little or no enzyme crossed the BBB and both IGF-II/MPR and MR uptake are blocked at that site. However, multiple high doses of GUS over time could partially clear storage in the brain of the MPS VII mouse. Because both high dose and longer circulation time were factors, we postulated that maintaining high levels of enzyme in the circulation for a protracted time was important. To test this hypothesis, we created an enzyme in which all carbohydrate-dependent, receptor-mediated uptake was inactivated by chemical modification with sodium meta-periodate. 11 This long-circulating form of GUS (PerT-GUS) was infused into MPS VII mice and found to be transported across the BBB.
GUS-GILT: Glycosylation-independent Lysosomal Targeting of GUS
Many strategies for treating lysosomal storage diseases are dependent on the delivery of enzyme via the M6P receptor. Consequently, most lysosomal enzymes used for therapy are expressed in mammalian cell lines, such as Chinese hamster ovary (CHO) cells, which contain the synthetic machinery for the addition of the M6P recognition signal to the oligosaccharides of the protein. However, some human lysosomal enzymes are poorly modified with M6P even in CHO cells. 12 Furthermore, the need for the M6P modification precludes the use of alternate expression systems such as bacteria, yeast, or insect cells, which cannot generate the M6P modification.
For this reason, with collaborators at Symbiontics, Inc. (now ZyStor, Inc.), we developed a peptide-based targeting system that was independent of the M6P modification to enable delivery of GUS. 8 We took advantage of the ability of the bifunctional IGF-II/MPR to bind the peptide hormone IGF-II. A portion of the IGF-II (hearafter referred to as the GILT tag) encoding residues 8–67 of mature human IGF-II was fused to the carboxyl terminus of human GUS, creating hGUS-GILT. This cassette was inserted into a mammalian expression vector and electroporated into CHO cells. After selection of a highly expressing CHO line, hGUS-GILT secreted from this line was collected, purified, and used for characterization and ERT studies.
The ability of GUS-GILT to be taken up by the M6P receptor was analyzed in an in vitro uptake system using human MPS VII fibroblasts. Figure 1A shows that uptake of untagged hGUS is inhibited completely by M6P, confirming that its uptake is entirely mediated by the M6P recognition marker. There is only partial inhibition by IGF-II, a phenomenon seen previously that has been attributed to steric inhibition and not competition for the M6P binding site. In contrast, hGUS-GILT is only partially inhibited by M6P, but more completely inhibited by IGF-II. This would indicate that its uptake is mediated by both M6P and the GILT tag. The hGUS-GILT that had been treated with endoglycosidase F1 to remove all M6P-containing oligosaccharides was taken up nearly as well as the untagged hGUS. Although it shows no inhibition by M6P, it is completely inhibited by IGF-II. These results indicate that the GILT tag alone is able to mediate binding and uptake by the bifunctional IGF-II/MPR through its IGF-II binding site.
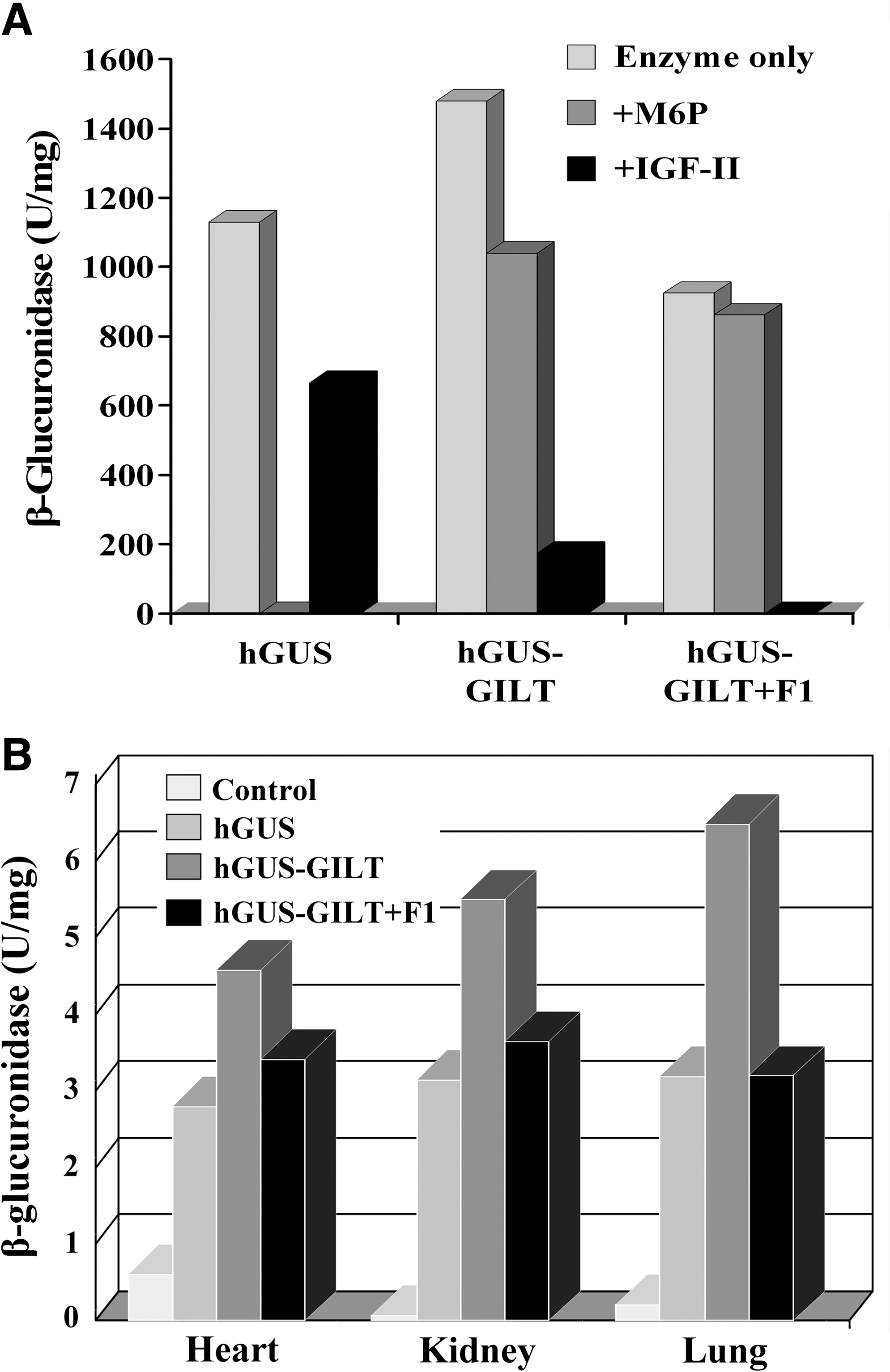
(
We next looked at the biodistribution of GUS, GUS-GILT, and GUS-GILT + F1 in MPS VII mouse tissues 24 h after infusion at a dose of 1 mg/kg. Figure 1B illustrates three tissues, heart, kidney, and lung, in which GUS-GILT is delivered at a higher level than GUS. It also shows that GUS-GILT + F1, devoid of M6P, is delivered to these tissues just as well as GUS. This result shows that the GILT tag by itself is able to deliver GUS-GILT to these tissues just as well as the M6P moeity on native GUS.
To test the relative effectiveness of GUS and GUS-GILT in reducing storage, MPS VII mice were infused with a short course of ERT consisting of three weekly treatments at a dose of 1 mg/kg body weight. It was evident that even with this short course of treatment, several tissues including liver were entirely cleared of storage. There were three notable differences in which GUS-GILT was more effective than GUS. GUS-GILT was able to reduce storage in the glomerular visceral epithelial (podocytes) and renal tubular cells in the kidney and in the osteoblasts in bone. Native GUS was less effective at removing storage at the three sites at the same dose (data not shown). 8
Together, these data suggest that use of the GILT tag on lysosomal enzymes could provide a “value added” effect in delivering enzyme to some sites more effectively than native enzyme. They also raise the possibility of using the GILT tag on nonlysosomal proteins or enzymes to mediate their delivery to lysosomes of cells that express the IGF-II/MPR.
GUS-Tat: Use of the 11-Amino Acid HIV Tat Peptide–GUS Fusion Protein to Facilitate Enzyme Delivery
The 11-amino-acid HIV Tat basic domain has been reported to have the ability to transduce denatured polypeptides from the extracellular space across the plasma membrane to the cytoplasm. 13 Once in the cytoplasm, these proteins are reported to refold into a functional form. Davidson et al. 14 reported the expression of a nondenatured GUS-Tat in an adenovirus vector, which led to wider distribution following intravenous or direct brain injection of the adenovirus vector. It was suggested that the additional distribution was due to the uptake of the enzyme by adjacent cells via adsorptive endocytosis mediated by the Tat domain. To see if purified protein would behave similarly, we expressed the GUS-Tat protein in CHO cells and purified it from the cell secretions. This purified chimeric enzyme in a nondenatured form was analyzed for its uptake properties and to see if the Tat tag facilitated the delivery of intravenously delivered enzyme during standard ERT. 9
We observed GUS-Tat uptake by human MPS VII fibroblasts that was only partially inhibited by M6P. The residual noninhibitable uptake was presumably mediated by the Tat moiety (data not shown). 9 To confirm this, we analyzed the portion of the uptake that was Tat mediated to see if it exhibited characteristics of adsorptive endocytosis. Figure 2A shows that increasing concentrations of negatively charged heparin completely inhibit the GUS-Tat uptake. Figure 2B shows that positively charged polylysine likewise inhibits GUS-Tat uptake in a concentration-dependent manner. Both of these compounds would inhibit the interaction between the Tat moiety on GUS-Tat and the heparan sulfate on the plasma membrane of the cell. This inhibition would prevent endocytosis of GUS-Tat during the normal internalization of the plasma membrane and, therefore, eventual delivery to the lysosome. We also observed that GUS-Tat uptake at 37°C was linear and nonsaturable with increasing concentrations of enzyme (data not shown). 9 These results, taken together, would suggest that GUS-Tat is, in fact, being taken up by absorptive endocytosis mediated by binding to heparan sulfate on the cell surface.
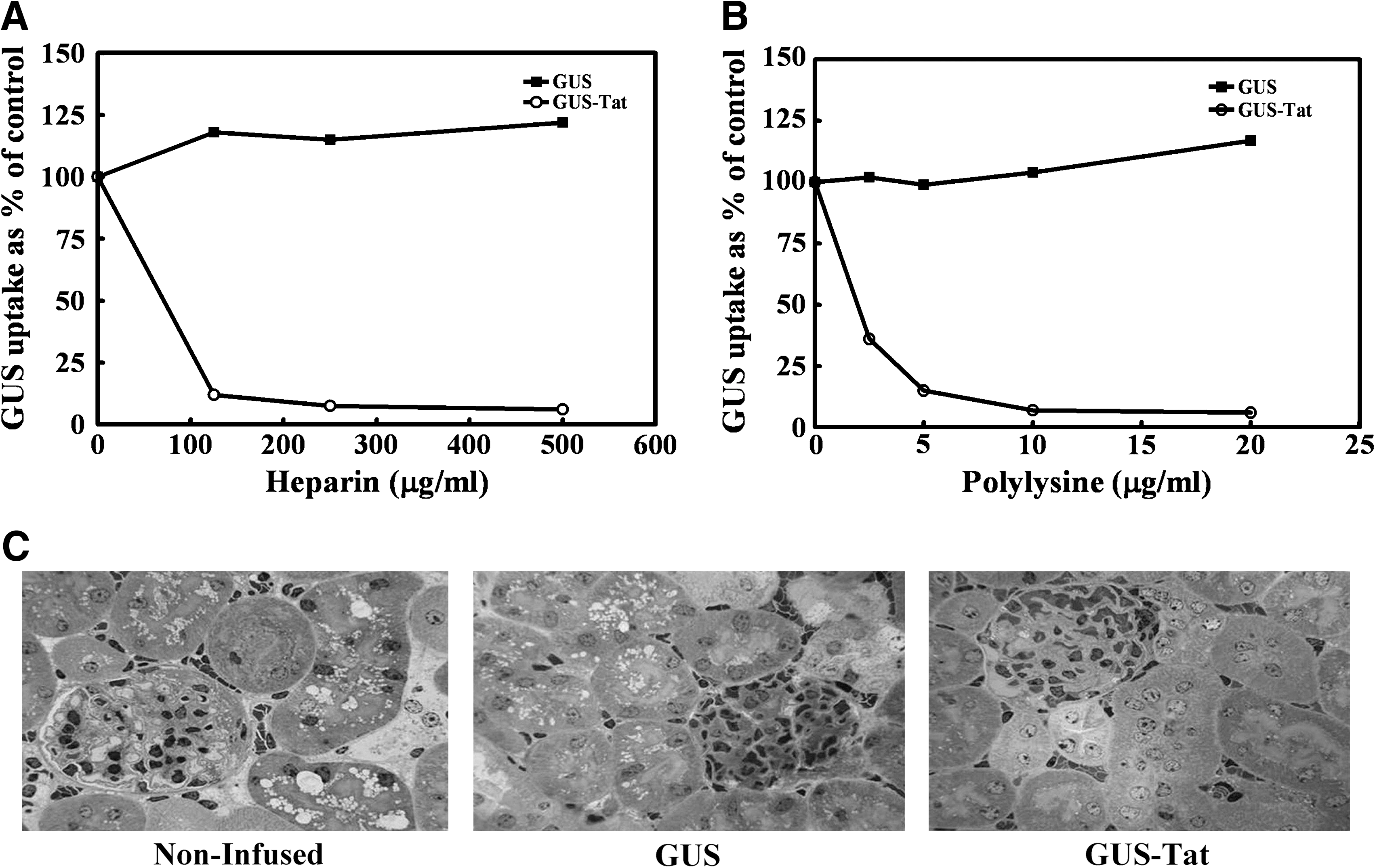
(
To see if GUS-Tat had any advantage over GUS for ERT, these compounds were compared using a protocol we had established previously. 8 This protocol consists of three weekly infusions of GUS or GUS-Tat (1 mg/kg) into the MPS VII mouse. One week after the last infusion, kidney was obtained at necropsy after perfusion and was fixed, sectioned, and stained with Toluidine Blue for evaluation of storage by light microscopy. Figure 2C illustrates the effectiveness of these infusions on clearance of storage in the renal tubular cells of the kidney. As can be seen in the first panel, there is abundant storage in the renal cells from noninfused animals. In the second panel, treatment with native GUS results in only partial correction of this storage, whereas in the third panel treatment with GUS-Tat resulted in almost complete correction. This improvement in the clearance of storage by GUS-Tat was also seen in the retinal pigment epithelium, bone osteoblasts, and osteoclasts, as well as in cardiac valve tissue (data not shown). 9
As we observed with the GILT tag, the Tat tag can mediate the delivery of lysosomal enzymes to the lysosome by an M6P-independent mechanism, in this case adsorptive endocytosis, and reach some tissues more effectively than native enzyme.
GUS-Fc: Use of the Fc Domain from Human IgG to Deliver GUS Across the Placenta
Recent evidence has shown that ERT for LSDs is most effective when initiated as early as possible. 15 In some LSDs, there are manifestations of the disease in the prenatal period. In fact, in MPS VII, some affected individuals present in utero with prenatal/neonatal hydrops, causing many of these infants to die in utero or within the first 2 years of life. 16 One way of treating these individuals prenatally would be to exploit a placental transport mechanism to deliver infused enzyme from the mother's circulation to the fetal circulation. One such transport system already exists to deliver IgG antibodies from mother to fetus. 17 IgG is delivered from the maternal circulation via interaction with the neonatal form of the FcRn. The FcRn binds the Fc domain on IgG in the maternal blood and mediates transcytosis across the syncytial trophoblast layer of the placenta. After release into the fetal circulation, the IgG is able to provide immunological protection to the fetus. A similar system using FcRn is also able transport IgG from maternal milk in the gut of the newborn across the intestinal epithelial cells into the newborn circulation. 18
We sought to exploit this process by the addition of the CH2-CH3 domain from the Fc portion of human IgG to the carboxyl terminus of the human GUS cDNA (GUS-Fc). 10 This fusion protein was expressed in CHO cells and purified from the conditioned medium from these cells. Purified GUS-Fc migrated as a 104-kD monomer on reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), which would be expected from adding the 29-kD Fc domain onto the 75-kD monomer of native GUS. To determine if GUS-Fc contained a functional Fc domain, we measured its ability to be precipitated by Protein G Sepharose. It was found that 74–85% of GUS-Fc was able to be precipitated by Protein G Sepharose as opposed to 5% of purified, untagged GUS (data not shown). 10 This result indicated that most of the GUS molecules contained functional Fc. However, on at least some molecules, the Fc domain had either been inactivated or removed proteolytically. Next, we looked at the susceptibility of GUS-Fc to be endocytosed by human fibroblasts and found that the uptake was reduced to 14% that of native GUS (data not shown). 10 This reduced rate of uptake of GUS-Fc was probably due to a lower M6P content. Similar reductions have been seen previously for other carboxy-terminal GUS fusion proteins, such as GUS-GILT and GUS-Tat.
To determine if GUS-Fc could be transported across the placenta, we measured the appearance of the enzyme in newborn (NB) pups of MPS VII mothers that had been intravenously infused two times with buffer only or with 380,000 units of GUS, GUS-Fc, or periodate-treated GUS (PerT-GUS) on embryonic days 17 and 18. These experiments were performed in both mannose receptor-positive and -negative mice. 19,20 Figure 3A shows that infusion of GUS produced no increase in the plasma levels of enzyme in the newborn pups of either type of mouse. In contrast, infusion of pregnant moms with GUS-Fc produced pups containing average plasma GUS levels of 2166 (MR+/+) units/mL or 4927 units/mL (MR−/−). As a control, we used PerT-GUS, a form of GUS devoid of both the M6P and mannose recognition signals that has an extremely long plasma t ½ of 18 h. Even with the extended time that PerT-GUS was present in the maternal plasma, there was essentially no transport across the placenta (Fig. 3A).

(
To provide further evidence that GUS-Fc is transported across the placenta by the FcRn, we used a genetic approach. We proposed that deleting either the gene for FcRn or β2-microglobulin (β2M) would prevent the transport of GUS-Fc across the placenta seen in C57/BL6 wild-type mice (B6). 21,22 To test this hypothesis, we used FcRn and the β2M knockout mice. 21,22 GUS-Fc was infused into pregnant females from B6 × B6, FcRn × FcRn, β2M × β2M, or FcRn × B6 matings on embryonic days 16 and 17. After birth, plasma GUS levels were assayed in 1-day-old pups. These infusions were performed in mice that were MPS VII+/+ and had endogenous levels of GUS in the plasma of 114 ± 14 units/mL. Figure 3B illustrates that GUS-Fc was detected in the plasma of pups produced by the B6 × B6 mating containing normal levels of FcRn. In contrast, no additional GUS over endogenous B6 levels was seen in the pups from both the FcRn × FcRn or the β2M × β2M matings. In addition, we observed only a 50% level of transport of GUS-Fc in pups from B6 × FcRn matings in which pups would be expected to have FcRn levels 50% of wild-type B6 mice.
To provide additional evidence that GUS-Fc transport was mediated by the FcRn, we studied a mutant form of GUS-Fc (GUS-Fc-TM) in which three amino acid residues (I, H, H) were substituted in the Fc domain. 23 These residues are known to be critical for binding of the Fc to the FcRn. When pregnant B6 mice were infused with GUS-Fc-TM, no pups exhibited plasma GUS levels greater than endogenous levels in pups from buffer-infused pregnant females.
To prove that the enzyme was transported prenatally, fetuses from pregnant females infused with GUS-Fc were removed by caesarean section on embryonic day 19. They contained even higher levels of GUS in their plasma than the 1-day-old pups. This finding ruled out the possibility that the pups were receiving the GUS-Fc only postnatally through the milk/gut route mentioned previously (data not shown). 11
PerT-GUS: Chemical Modification of Carbohydrate to Create a Long Circulating Form of GUS
One of the unmet needs of ERT for the lysosomal storage diseases is a strategy to deliver infused enzyme to the brain and other areas of the central nervous system (CNS). 24 Lack of this delivery has compromised the effective clearance of storage in the lysosomes of these tissues. One of the factors is the rapid clearance of enzyme by high levels of mannose and M6P receptors in organs such as the liver and spleen. These organs remove enzyme after infusion so quickly that other organs, including brain, receive a rather small percentage of the infused enzyme. Recently, several studies showed that multiple infusions of high doses of enzyme result in partial clearance of storage in the brain. 25 –28 What these studies have in common are conditions in which relatively high levels of enzyme remained in the blood for prolonged periods of time. Therefore, we proposed that this delivery might be due to exposure of the brain capillaries to repeated high levels of enzyme in the circulation.
Earlier studies had shown that carbohydrate-mediated clearance (clearance by the galactose, mannose, and M6P receptors) could be eliminated by periodate modification of the carbohydrates on lysosomal enzymes. 29 –31 To determine if time of exposure to brain capillaries itself was important, this method (chemical inactivation with sodium meta-periodate) was used to make a form of GUS (PerT-GUS) that was extremely long circulating. It was necessary to follow this reaction by borohydride reduction to block the reactive aldehydes produced in the periodate reaction and to prevent crosslinking of the protein.
To confirm that the carbohydrate had been inactivated effectively, PerT-GUS was tested for its ability to be taken up by M6P receptors on human fibroblasts or by mannose receptors on mouse macrophages. We observed that the enzyme uptake in these cell lines by each of these receptors had been completely inactivated (data not shown). 11 To determine the effect of the periodate treatment on plasma clearance, mice were infused with GUS or PerT-GUS at a dose of 4 mg/kg body weight. After infusion, blood samples were taken at intervals and assayed for plasma GUS activity. Figure 4A shows that GUS is cleared rapidly from the plasma with a t ½ of 11.7 min. In contrast, the clearance of PerT-GUS was dramatically prolonged to a t½ of 18.5 h. These studies indicate that plasma clearance by the carbohydrate receptors has been successfully abrogated by the periodate treatment.
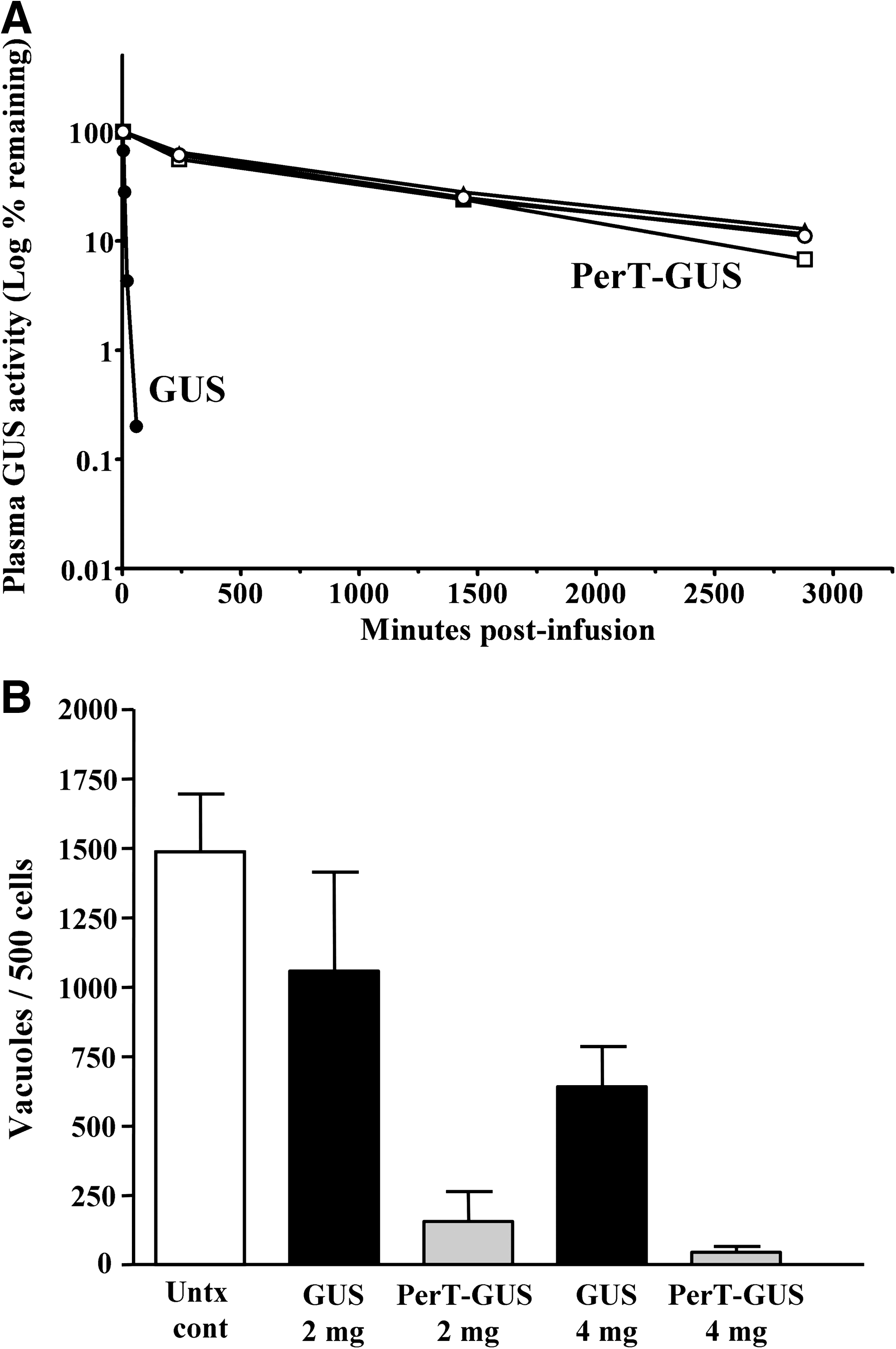
(
The tissue distribution of GUS vs. PerT-GUS was determined by intravenously infusing MPS VII mice one time with a dose of 4 mg/kg. Forty eight hours after infusion, the mice were perfused and organs were collected for GUS assay. We found that PerT-GUS was delivered to all tissues assayed (data not shown). 11 In the liver and spleen, tissues usually responsible for substantial uptake of GUS due to endogenous mannose and M6P receptors, PerT-GUS levels were actually reduced to about 25% of the levels obtained by infused GUS. However, in heart, kidney, lung, muscle, and eye, PerT-GUS was delivered to levels 2- to 5-fold those of infused GUS. Most importantly for these studies, PerT-GUS was delivered to the brain, attaining levels of 1.30 ± 0.28 units/mg. This is 6 times the level achieved with native GUS (0.23 ± 0.005 units/mg). This level of PerT-GUS achieved in the brain is 7.8% of wild type (16.7 ± 2 units/mg), which is almost certainly high enough to effect clearance of storage.
To assess the effectiveness of PerT-GUS in clearing neuronal storage, MPS VII mice tolerant to human GUS were infused weekly times 12 weeks by tail vein infusion with doses of 2 or 4 mg/kg body weight with GUS or PerT-GUS. One week after the last infusion, brain tissue was obtained at necropsy after perfusion and was fixed, sectioned, and stained for evaluation of storage by light microscopy. We observed in neocortical and hippocampal neurons that treatment with GUS was only partially effective at both the 2-mg and 4-mg doses (data not shown). 11 PerT-GUS, in contrast, affected almost complete clearance of storage at these locations (data not shown). 11 To obtain a more objective measure of the reduction in neuronal storage, we devised a morphometric method. In this method, we counted the actual number of storage vacuoles in 500 contiguous neurons. Figure 4B shows the results of the morphometric analysis in neocortical neurons. It is evident that treatment with GUS at either the 2-mg/kg or 4-mg/kg doses resulted in only a partial clearance of storage. On the contrary, it can be seen that PerT-GUS at either dose produces almost complete reversal of storage. We likewise saw with both doses almost complete reduction of storage by PerT-GUS in hippocampal neurons (data not shown). 11
The exact mechanism that enables PerT-GUS to cross the BBB still needs to be elucidated. Possible mechanisms are: (1) Binding to a low-affinity receptor that is only unmasked when the enzyme is not rapidly cleared by the IGF-II/MPR and MR clearance systems; (2) PerT-GUS is transported across the BBB by fluid-phase pinocytosis; (3) modification of GUS to create a ligand now recognized by a cryptic receptor in brain capillaries; or (4) uptake of PerT-GUS by circulating macrophages that carry it across the BBB. These studies, showing clearance of brain storage by PerT-modified enzyme, are very provocative and may hold great promise.
Summary
We have presented several alternative ways to deliver lysosomal enzymes to the lysosome to address unmet needs. We have shown that novel tags such as GILT or Tat can be used to augment or replace normal delivery by the mannose and M6P receptors. In another application, we found the use of the Fc tag can be used to deliver a lysosomal enzyme across the placenta to the growing fetus, a route not available to native enzyme. Chemically modified PerT-GUS surprised us and suggested the advantages of using an enzyme that is apparently devoid of receptor recognition signals. Defining the mechanism by which it reaches resistant sites, including the brain, and exploring its generality are current challenges. The payoff could be huge.
In the field of bioremediation, studies have shown that compounds can accumulate in lysosomes over time that cannot be broken down by the natural contingent of lysosomal enzymes. 32 These accumulated compounds can lead to lysosomal dysfunction associated with aging or other pathologies. One example is the build up of 7-ketocholesterol in the lysosomes of macrophages during atherosclerosis, disrupting their entire cholesterol-processing machinery. Recent studies have identified enzymes from the biome that have the potential to break down the 7-ketocholesterol to reverse this disruption. 33 Strategies will be needed to deliver this type of nonlysosomal enzyme to the lysosome. The types of tags described here could be easily incorporated into the protein sequence of newly identified enzymes from the biome to direct them to the lysosome.
Footnotes
Acknowledgments
We thank Tracey Baird for excellent editorial assistance on this manuscript. This work was supported by National Institutes of Health grant GM34182 to W.S.S.