
Abstract
Protein aggregation occurs in many age-related neurodegenerative diseases, where it can lead to deposits of naturally occurring proteins in the brain. In case of Creutzfeldt–Jakob disease (CJD), these deposits consist of prion protein (PrP). CJD has three etiologies: spontaneous, genetic, or caused by infection. A polymorphism within the PrP gene is associated with susceptibility of infection. The main event in prion diseases is the conversion of PrP from its naturally occurring isoform to its disease-associated isoform. Here, we present the adaption of a previously reported in vitro conversion system based on hamster recombinant PrP to analyze amyloid fibril formation of human recombinant PrP. We further compare the aggregation characteristics of the human PrP according to the polymorphism variants M129 and V129.
Introduction
CJD occurs in three etiologies: spontaneous, genetic, or caused by infection. The median age of death for the spontaneous case of CJD (sCJD) is 68 years. 1 The incidence of sCJD decreases at the age of more than 79. 2 In contrast, individuals that are afflicted by variant CJD (vCJD), which is connected to the consumption of bovine spongiform encephalopathy (BSE)-contaminated meat and therefore is discussed as an infectious case, have a median age of death of 28 years. 1 In this context, why sCJD is age-related, whereas vCJD occurs in younger individuals, is a challenging question. 3 The amino acid replacement M129V according to the polymorphism of the PrP coding gene seems to be a relevant factor for the incidence of CJD. The M129 variant is associated with susceptibility of infection because the documented vCJD cases are homozygous for methionine at amino acid position 129. 4 The M129 variant also dominates sCJD. A total of 72% of all documented sCJD cases have this genotype. 5
The key molecular event in prion diseases is the conversion of PrP from its naturally occurring isoform (PrPC) to its disease-associated isoform (PrPSc). PrPC is noninfectious and characterized by solubility in mild detergents, an α-helical–rich secondary structure and sensitivity against proteinase K (PK) digestion, whereas the disease-associated isoform PrPSc is infectious, insoluble, β-sheet–rich in secondary structure, and partially resistant against PK digestion. 6
Previously, we reported a PrP in vitro conversion system that allows the analysis of the mechanism underlying aggregation and formation of amyloid fibrils from soluble PrP. 7 –9 This assay was based on hamster recombinant PrP in solution with well-balanced salt and sodium dodecyl sulfate (SDS) concentrations, providing a capable environment for fibrillization of PrP. 10 In the present study, we adapted the assay to use full-length human recombinant PrP (hu recPrP) and compared hu recPrP with different amino acids at position 129 with respect to its conversion characteristics.
Materials and Methods
recPrP was expressed in Escherichia coli. Therefore, the PrP sequence (coding for amino acids 23–230) was cloned in a pET-11a vector, and the resulting plasmid was transformed in Rosetta 2 (DE3) (Merck KGaA). According to the polymorphism at amino acid 129, the two following constructs were expressed: (1) hu recPrP, amino acids 23–230 with methionine as amino acid 129, referred to as “hu recPrP M129,” and (2) hu recPrP, amino acids 23–230 with valine as amino acid 129, referred to as “hu recPrP V129.” recPrP was expressed and purified as described in ref. 7. After purification, hu recPrP was dissolved in 10 mM NaPi and 0.2% SDS. Under these conditions, hu recPrP is soluble, monomeric, and α-helical in secondary structure. To analyze its aggregation, hu recPrP was diluted to a final concentration of 300 ng/μl within the in vitro conversion system. Samples were finally buffered by 10 mM sodium phosphate, pH 7.4 (NaPi), 250 mM NaCl, and 0.02% SDS (referred to as conversion buffer) in a total volume of 250 μl.
The analysis of the preamyloid state by circular dichroism (CD) and analytical ultracentrifugation as well as the characterization of the amyloid amount of aggregates by thioflavin T (ThT)-assay, Congo Red staining, and transmission electron microscopy (TEM) were carried out as described in refs. 7 and 8.
Results
Previously, we established an in vitro conversion system for recPrP, in which recPrP first had many characteristics of PrPC and then after the conversion process had many characteristics of PrPSc. Because the conversion system is based on well-balanced concentrations of SDS and salt, we established buffer conditions, in which human recPrP aggregates into fibrils by varying the SDS concentrations. Suitable conditions for amyloid fibril formation of hu recPrP were 0.02% SDS in 10 mM NaPi and 250 mM NaCl. Thus, these buffer conditions (conversion buffer) were used in all further experiments.
To characterize hu recPrP precursor state before fibril formation (preamyloid state), the secondary structure was analyzed by CD spectroscopy. The spectra were measured immediately after addition of hu recPrP to the conversion buffer and additionally after 24, 48, and 72 h. The secondary structures of hu recPrP M129 and V129 were compared. All spectra showed an α-helically dominated secondary structure, based on minima at 208 and 222 nm, with parts of random coil structure, indicated by the distinctive minima at 208 nm (Fig. 1A,B). In contrast to hu recPrP V129, the M129 variant showed a faster decrease in the CD amplitudes of the spectra, which could be explained by protein aggregation (Fig. 1A). Spectra measured using a mixture of both variants of hu recPrP as a model for the heterozygous case showed a linear combination of the spectra of both variants alone (data not shown). The preamyloid state was also analyzed by analytical ultracentrifugation. It revealed the presence of a dimer for both PrP variants M129 and V129 (data not shown).
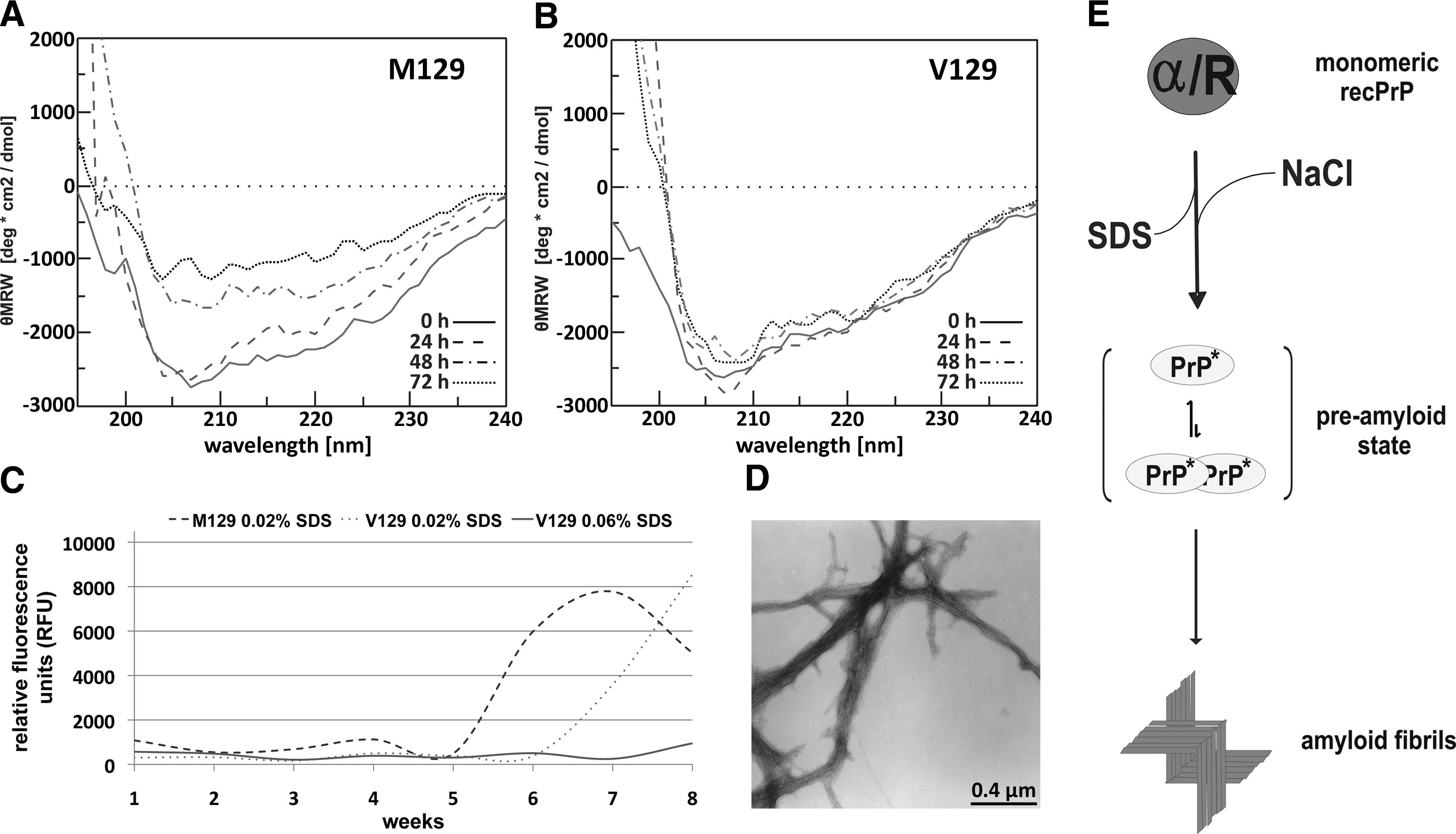
(A, B) Secondary structure analysis of the preamyloid state of human recombinant prion protein (hu recPrP) (300 ng/μl) in conversion buffer obtained by circular dichroism spectroscopy after 0, 24, 48, and 72 h. (A) hu rec PrP M129. (B) hu recPrP V129. (C) Kinetics of amyloid formation of hu recPrP shown by thioflavin T (ThT) time-dependent fluorescence. The kinetics of amyloid formation of hu recPrP (300 ng/μl) M129 and V129 in conversion buffer and in similar buffer with 0.06% sodium dodecyl sulfate (SDS) as control are shown. (D) Ultrastructure of hu recPrP M129 was analyzed by transmission electron microscopy. Bar equals 0.4 μm. (E) Schematic model of the in vitro conversion mechanism. The monomer has an α/random secondary structure. By diluting the SDS concentration and adding NaCl, the protein forms amyloid fibrils via a monomer/dimer preamyloid state.
To assay PrP aggregates for their amyloid content, ThT fluorescence was used. After 6 weeks of incubation at 37°C and 650 rpm, hu recPrP M129 showed an amyloid-content dependent increase of ThT fluorescence (Fig. 1C). After 7 weeks of incubation, the hu recPrP V129 showed a substantial increase of ThT-dependent fluorescence (Fig. 1C). The control (hu recPrP in 10 mM NaPi, pH 7.4, 250 mM NaCl, 0.06% SDS) did not show any increase of fluorescence intensity within the time frame of the measurement. After the hu recPrP sample reached saturation of ThT fluorescence, the fluorescence intensity started to oscillate heavily, probably as a result of the assay setup, e.g., larger PrP aggregates sank to the bottom and were resuspended again.
Thus, from our data, one may conclude that the M129 PrP variant aggregates faster than the V129 variant. This is in contrast to results of the Baskakov group, who reported the V129 to aggregate faster than the M129 variant. They used a different conversion system based on guanidinium chloride and urea to induce fibrillization of the amino-terminally truncated form hu recPrP (amino acids 90–231). 11 This difference can either be ascribed to the different conversion conditions or, more likely, to the use of non–full-length prion proteins.
The amyloid hu recPrP aggregates produced within the conversion system were additionally analyzed by Congo Red staining. For hu recPrP M129 and V129, the presence of amyloids could be confirmed (data not shown). Samples that did not show an increase of fluorescence intensity in the ThT assay, did not show amyloid-specific binding of Congo Red (data not shown).
To determine the ultrastructure of the amyloid aggregates, we performed transmission electron microscopy of the ThT- and Congo Red-positive hu recPrP. Both hu recPrP M129 and V129 revealed a fibrillar ultra structure (Fig. 1D).
Conclusions and Outlook
In summary, we were able to generate amyloid fibrils from both human prion protein variants with methionine or valine at residue position 129 using the previously described in vitro conversion system. 7 –10 We characterized the amyloid precursor state of hu recPrP and propose an α-helical dimer as an intermediate state before fibrillization. The preamyloid states of both M129 and V129 variants show virtually identical properties: soluble dimers with α-helical secondary structure. The aggregation kinetics of both variants, however, differed. hu recPrP M129 aggregated faster than the V129 variant. Anticipating that a higher aggregation tendency for M129 leads to increased risk of disease incidence, these in vitro data are in intriguing accordance with observed disease phenomena of CJD in vivo, because the homozygous phenotype M129 dominates sCJD cases and is exclusively found in vCJD cases. Thus, aggregation kinetics of PrP variants may well be a relevant, if not decisive, factor of CJD incidence.
Footnotes
Acknowledgments
This work was supported by the DFG Research Training Group 1033, Molecular Targets of the Aging Process and Strategies for the Prevention of Aging, the Network of Excellence Neuroprion, the Helmholtz-funded virtual institute VIBS (VH-VI-013), and the Food Standards Agency, UK.
Author disclosure statement
No competing financial interests exist.