
Abstract
Previous studies examining the association between the glutathione S-transferase omega-1 (GSTO1) single-nucleotide polymorphisms (SNPs) and Alzheimer disease (AD) have yielded conflicting results. Furthermore, an effect of GSTO1 rs4925 on the age-at-onset (AAO) of AD was found in different studies on sporadic and familial AD cases, but with contrasting findings. A total sample of 103 AD patients, and 157 age- and sex-matched unrelated caregivers from Apulia, southern Italy, were genotyped for the apolipoprotein E (APOE) polymorphism and the GSTO1 rs4925 and rs1804834 SNPs. Furthermore, we performed a haplotype analysis on these two SNPs on the GSTO1 locus and evaluated the possibility of interaction with APOE. Significant differences were observed in rs4925 genotype distribution between AD patients and age- and sex-matched healthy controls. Both the C/A (odds ratio [OR] = 3.116; 95% confidence interval [CI], 1.749–5.550) and the A/A (OR = 10.802; 95% CI, 3.605–32.128) genotypes resulted in an association with AD. A higher frequency of the allele A was observed in AD patients than in age- and sex-matched controls (OR = 3.789; 95% CI, 2.442–5.878). No significant differences were observed in the rs1804834 genotype or allele frequencies between AD patients and controls. No significant influence of the GSTO1 genotypes on the AAO was observed. No significant interaction was found among the GSTO1 SNPs and APOE. In both AD and controls, no important linkage disequilibrium (LD) was observed among the markers investigated. Whereas the C-A haplotype appeared to be protective against AD (OR = 0.303; 95% CI, 0.204–0.451), the A-A haplotype appeared to be at increased risk for AD (OR = 4.014,; 95% CI, 2.528–6.382). Our findings supported a role of the GSTO1 rs4925 SNP in the risk of sporadic AD in southern Italy, suggesting that this and other variants of the GSTO1 gene could be implicated in AD pathogenesis.
Introduction
Studies on the biologic functions of GSTO1 have been rare. Ubiquitous expression of GSTO1 suggested a function as a housekeeping gene, possibly protecting from oxidative stress because of its dehydroascorbate reductase and thiol transferase activities; furthermore, GSTO1 may have an important role as a nuclear antioxidant system. 1 GSTO1 may contribute to the AD pathogenesis by affecting the cellular redox homeostasis or by modifying the posttranslational processing of interleukin-1β (IL-1β. 13 The transcripts of GSTO1 were also significantly downregulated in the hippocampus obtained from AD patients compared with that of controls. 14 In humans, the GSTO1 gene is located on the long arm of chromosome 10, at locus 10q25.1. This gene has two polymorphism in exon 4: rs4925 (C428 → A), determining the amino acid change Ala140 → Asp, and the silent rs1804834 (A441 → G). Human variants of GSTO1, which reduced enzyme activity and therefore might influence the susceptibility to oxidative stress, have been characterized. 15 Some recent studies have demonstrated that polymorphisms in the GSTO1 gene may have relevance in AD, vascular dementia, and stroke. 14,16 In particular, several studies conducted on European, Asiatic, and U.S. populations to verify the presence of an association among GSTO1 polymorphisms and the risk of AD have found contrasting findings. 16 –25 In line with these suggestions, a recent study described that the GSTO1 locus might influence the age-at-onset (AAO) of AD and Parkinson disease (PD). 19 In fact, an effect of GSTO1 rs4925 on the AAO of AD was found in different studies on sporadic and familial AD cases, but with contrasting findings. 16 –20,24
In the present study, our first aim was to test the GSTO1 rs4925 and rs1804834 SNPs for an association with the disease in a sample of sporadic AD patients and age- and sex-matched control subjects from southern Italy. Second, we performed a haplotype analysis on these two polymorphisms to examine possible linkage disequilibrium (LD). Finally, we evaluated the possibility of interaction of these two GSTO1 SNPs with APOE.
Materials and Methods
Subjects
A total sample of 260 subjects from Apulia (southern Italy) was studied. All subjects included in the present study were Caucasians, born and residing in southern Italy. All four grandparents of all subjects included in the present study were born in Italy. The AD group consisted of 103 patients (62 women and 41 men, mean age at onset 71.2 ± 10 years) including 75 patients with sporadic late-onset AD (age at onset ≥65 years, 46 women and 29 men, mean actual age 76.3 ± 5.6 years), and 28 patients with sporadic early-onset AD (age at onset <65 years, 16 women and 12 men, mean age at onset 57.5 ± 4.2 years), consecutively examined between June, 2002, and April, 2008, at the Centre for Aging Brain, Memory Unit, Department of Geriatrics, Bari University Hospital, Italy. This center is the largest clinical setting for AD diagnosis in our region.
Clinical diagnosis of probable AD was made according to the National Institute for Neurological and Communicative Disorders and Stroke/Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA) criteria. 26 The AAO of AD symptoms was estimated by semistructured interviews with the patients' caregivers. 27 Cognitive status (orientation, attention, immediate and delayed verbal memory, constructional praxis, and language) was screened in all patients by means of the Mini-Mental State Examination (MMSE). 28 Functional status was assessed with the Activities of Daily Living (ADL) scale (scores ranging from 6 [all functions preserved] to 18 [all functions lost]), which determines the level of independence in six activities: Bathing, dressing, toileting, transferring from bed to chair, continence, and feeding. 29 Ability in home management was assessed by the Instrumental Activities of Daily Living (IADL) scale (scores ranging from 8 [all functions preserved] to 31 [all functions lost]), which determines the level of independence in executing tasks such as using the telephone, shopping for personal items, preparing meals, doing light housework (e.g., washing dishes), managing money or drugs, and so forth. 30 To be eligible for inclusion in this study, patients were required to have a Clinical Dementia Rating (CDR) scale (range, 0–5) score of 0.5 or higher, 31 a modified Hachinski ischemic score (range, 0–12) less than 3, 32 and a Hamilton depression scale (range, 0–67) score less than 17. 33
The nondemented age-, sex-, and ethnically matched control group included 157 unrelated caregivers (79 women and 78 men, mean age 70.8 ± 16.4 years) spouses, friends, neighbors, or volunteers, consecutively examined from between 2002 and April, 2008. This group included 131 subjects with a mean actual age of 77.0 ± 5.3 years (57 women and 74 men), and 26 subjects with a mean actual age of 37.7 ± 12.6 years (22 women and 4 men), with no known diagnosis of dementia or other chronic neurological diseases or psychiatric syndromes with cognitive impairment, cerebrovascular disease, nephropathy or end-stage renal disease, and no severe functional limitations. The ascertainment, diagnosis, and collection of cases and controls has been described in detail elsewhere. 27 After a complete description of the study, written informed consent was obtained from all subjects and/or their relatives prior to collecting of blood sample. The study protocol received the approval from the Ethical Committee of the University of Bari.
Genetic analysis
Genomic DNA was extracted from peripheral blood samples by the GFX Genomic Blood DNA Purification Kit (Amersham Biosciences). APOE genotypes were determined as detailed elsewhere. 34 After careful computer-aided analysis of the target sequence and to avoid mishybridization, rs4925 genotyping was performed on a Light Cycler system (Roche, Mannheim Germany) by melting curve analysis using a specifically designed hybridization probe with fluorescent dyes: Fluorescein isothiocyanate (FITC)-labeled sensor (5′-CTTCTTTTAGGCCAGCATAGTCTTCTTTA—FL-3′) and Light Cycler (LC) Red 640 Anchor (5′-TGGCTTCTAATAAAGCTTCCTACCAAGGATGGC—PH-3′; TIBMol Biol) probes. Also, rs1804834 genotyping was performed on a Light Cycler system by melting curve analysis using specifically designed hybridization probe with fluorescent dyes: FITC-labeled sensor (5′-ACCAAGCTAGAGGAGGTAATTATTTCTCCT—FL-3′). For the anchor probes to type the 3-bp deletion downstream, we had to use Locked Nucleic Acid (LNA) bases to gain a sufficient melting temperature for this probe as follows: Light Cycler (LC) Red 705 Anchor (5′-GCTALNATCALNATCALNAGALNAGTALNAAACGALNAT AALNACTALNATATC—PH-3′; TIBMol Biol) probes. The primer used in the amplification were: forward primer, 5′-GACCAAGCCAGCATTTTAGG-3′; reverse primer, 5′-AGC AAGCCCATGACAAAGTCT-3′.
The following 20-mL reaction mix was used for PCR amplification and subsequent detection of fluorescence from hybridization probes: 100 ng of genomic DNA, 50 pmol each primer, 3 pmol each probe, and 3.5 mM MgCl2. A PCR “touchdown” was performed to reduce mispriming and increase efficiency. The amplification program consisted of initial denaturation of 2 min at 95°C, then 45 cycles with three temperature segments. The first segment was 94°C for 10 sec at 20°C/s of temperature transition rate for denaturation; a second segment for both primer and probe annealing was of 15 sec at 62°C for five cycles followed by a 0.4°C decrease of the annealing temperature every second cycle up to 55°C; the temperature transition rate was 20°C/sec. A third temperature segment for primer extension was 72°C for 10 sec at 20°C/sec of temperature transition rate. After amplification, the melting curve analysis program was performed with three different temperature segments, as well: The temperature was initially raised to 94°C for 30 sec for denaturation, then lowered to 40°C at 20°C/sec, and held at 40°C for 1 min to permit probe annealing, and finally raised to 90°C at 0.15°C/sec of temperature transition rate while collecting fluorescence data continuously to obtain the melting curve analysis profile temperature. A final cooling at 40°C was carried out. The melting temperatures were 64°C for A allele, 66°C for C allele, 59°C for D allele, and 67°C for I allele.
Statistical analysis
Differences in age and education between the AD group and age- and sex-matched healthy controls were analyzed by the Student t-test for independent data, whereas differences in sex were evaluated by the Pearson chi-squared test. Differences in MMSE score, ADL score, and IADL score between AD group and age- and sex-matched healthy controls were analyzed by Mann–Whitney U-test. The Hardy–Weinberg equilibrium was tested for both AD and controls. A two-tailed Pearson chi-squared test was used to compare genotype frequency distribution between AD patients and controls (n × 2 contingency tables). These tests were performed using the R Ver. 2.8.1 statistical software package (The R Project for Statistical Computing; available at
The relative allelic frequencies were estimated by the gene-counting method.
35
To express variances of genotype and allele frequencies we used 95% confidence intervals (CI), the upper and lower values of which were calculated according to the Wilson formulas. The Fisher exact test was used to estimate the odds ratios (ORs) in testing for possible association between the single genotypes/alleles/haplotypes and AD (2 × 2 contingency tables), and was performed using the 2-Way Contingency Table Analysis available at The Interactive Statistical Calculation Pages (available at
Results
Sociodemographic and clinical characteristics of AD patients and age- and sex-matched healthy controls are summarized in Table 1. Genotype and estimated allele frequency distributions for the investigated polymorphisms are summarized in Table 2. Significant differences were observed in rs4925 genotype distribution between AD patients and age- and sex-matched healthy controls. Assuming as the reference the wild-type C/C genotype, a higher frequency of the C/A genotype (36.89% vs. 19.75%, p < 0.001) and of the A/A genotype (16.50% vs. 2.55%, p < 0.001) was observed in AD patients than in controls. Thus, both the C/A (OR = 3.116; 95% CI, 1.749–5.550) and the A/A (OR = 10.802; 95% CI, 3.605–32.128) genotypes were associated with AD. As expected from these genotype frequencies, a higher frequency of the allele A (0.350 vs. 0.124, p < 0.001) was observed in AD patients than in age- and sex-matched controls. Therefore, the allele A (OR = 3.789; 95% CI, 2.442–5.878) was associated with AD. No significant differences were observed in rs1804834 genotype frequencies between AD patients and controls. Assuming the wild-type A/A genotype as the reference, no significant differences were observed in the distribution of the A/G (p = 0.385) or the G/G (p = 0.999) genotype. As expected from these genotype frequencies, no difference was observed in the distribution of the G allele (p = 0.340). We also investigated the possible influence of the GSTO1 genotypes on the AAO. No significant influence was observed (data not showed).
Values are expressed as mean ± standard deviation (SD) unless otherwise indicated.
Evaluated by Student t-test for independent data.
Evaluated by Pearson chi-squared test.
Evaluated by Mann–Whitney U-test.
M, Male; F, female.
Hardy–Weinberg equilibrium:
AD: p = 0.079 (rs4925), p = 1.000 (rs1804834), and p = 0.368 (APOE);
Controls: p = 0.148 (rs4925), p = 1.000 (rs1804834), and p = 1.000 (APOE).
95% CI of the estimated genotype frequencies:
rs4925 in AD: 0.39–0.55 (C/C), 0.29–0.45 (C/A), and 0.10–0.22 (A/A);
rs4925 in controls: 0.71 - 0.85 (C/C), 0.14–0.26 (C/A), and 0.00–0.04 (A/A).
rs1804834 in AD: 0.90 - 0.72 (A/A), 0.27 - 0.09 (A/G), and 0.03 - 0.00 (G/G)
rs1804834 in controls: 0.93 - 0.77 (A/A), 0.22 - 0.06 (A/G), and 0.03 - 0.00 (G/G)
95% CI of the estimated allele frequencies
rs4925 in AD: 0.70 - 0.60 (C), and 0.40 - 0.30 (A)
rs4925 in controls: 0.91 - 0.85 (C), and 0.15 - 0.09 (A)
rs1804834 in AD: 0.93 - 0.87 (A), and 0.13 - 0.07 (G)
rs1804834 in controls: 0.95 - 0.89 (A), and 0.11 - 0.05 (G)
SNP ID, Single-nucleotide polymorphism identification; OR, odds ratio; CI, confidence interval; h, effect-size; PWR, statistical power.
The statistical models to investigate the possible synergy between GSTO1 SNPs and APOE polymorphism are summarized in Table 3. In these models, considering each GSTO1 SNP, no significant interactions were found. Similarly, in the model considering both GSTO1 SNPs, no synergy was also found. Furthermore, the estimated haplotype frequencies at GSTO1 locus in AD patients and controls are summarized in Table 4. Haplotype C-A was the most frequent (57.7% in AD patients and 82.0% in controls), followed by the A-A haplotype (32.1% in AD patients and 10.4% in controls) and by the C-G and A-G haplotypes (frequency <10% in both AD and controls). In both AD and controls, no important LD was observed among the markers investigated, i.e., D′ = 1.000 (r 2 = 0.061) and D′ = 0.195 (r 2 = 0.022), respectively, for AD and controls (Fig. 1). Whereas the C-A haplotype appeared to be protective against AD (OR = 0.303; 95% CI, 0.204–0.451), the A-A haplotype appeared to be at increased risk for AD (OR = 4.014; 95% CI, 2.528–6.382).
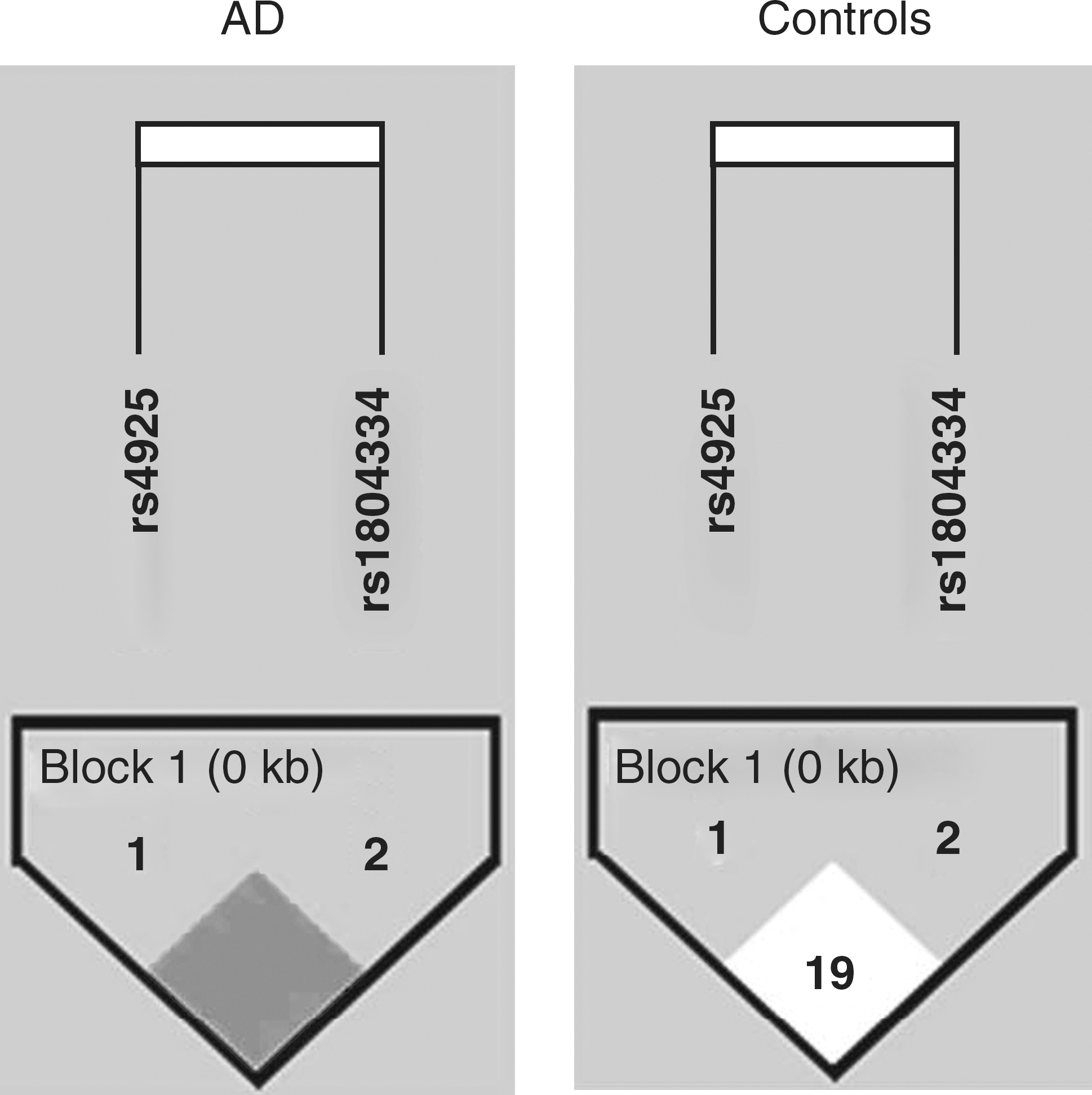
Schematic representation of linkage disequilibrium (D′ coefficient) among the markers rs4925 and rs1804334 at the glutathione S-transferase omega-1 (GSTO1) gene locus in Alzheimer disease (AD) patients and nondemented age- and sex-matched controls.
df, Degrees of freedom.
OR, Odds ratio; CI, confidence interval; h, effect-size; PWR, statistical power.
Discussion
In the present study, we reported that the GSTO1 rs4925 genotypes were variously associated with AD, with the rs4925 A allele increasing the risk of the disease. Furthermore, the C-A haplotype appeared to be protective against AD, whereas the A-A haplotype showed an increased risk of AD. However, in both AD and controls, no important LD was observed among the markers investigated.
Only a few studies on GSTO1 polymorphisms have shown a weak influence on the risk for AD, 17,18 whereas other reports did not find any association with the disease. 16,19 –25 To the best of our knowledge, this study is the first to link an increased risk of sporadic AD to the GSTO1 rs4925. In fact, of the two studies that suggested a role of rs4925 in the risk of AD, one report found a weak allelic association with the AD phenotype 17 in a unique cohort of families of Caribbean Hispanic ancestry and the other report revealed only a modest association of one three-site [GSTO1 and GSTO2 genes and the protease serine 11 (PRSS11) gene] haplotype with AD risk. 18
In the present study, we were unable to replicate the reported association of GSTO1/GSTO2 SNPs with AAO in AD. 19 Although Li and colleagues confirmed their original findings demonstrating that the delay of AAO by a GSTO1–GSTO2 haplotype can be as long as 13 years in both AD and PD, 24 many other studies did not find an influence of the SNPs in GSTO1 and GSTO2 on AAO in AD. 17,18,20 Furthermore, Kölsch and colleagues have confirmed an association of GSTO1 with AAO in AD, 16 as well as vascular dementia (VaD) and stroke, although with the opposite allele for late AAO to the one that was originally reported by Li and colleagues. 19 Given that Li and colleagues used familial AD cases in their studies, 19,24 it is also possible that the effect of these genes is stronger in familial AD due to shared common genetic background than in sporadic AD, thus making it difficult to readily detect it in sporadic AD cases.
In the present study, we estimated the power of the haplotype analysis at ≈100%. This value is clearly overestimated. However, the difference in the proportion of haplotypes between AD cases and controls lead to an effect size of −0.601 (haplotype C-A) and 0.615 (haplotype A-A). Moreover, the haplotype associations showed a small CI. This suggested that the associations perhaps are not spurious, which is also confirmed by the presence of haplotypes with an opposite effect toward AD.
The mechanisms underlying the possible association of the GSTO1 locus with sporadic AD are, at present, unclear. GSTO1 is a stress response protein, and it may contribute to the AD pathogenesis given its role in cellular redox homeostasis. 1 In fact, rs4925 has been associated with lower thioltransferase activity and therefore may influence the intracellular thiol status, explaining the variation between individuals in their susceptibility to oxidative stress. 15 GST activity has been shown to be decreased in the AD amygdala, hippocampus, parietal lobe, and nucleus basalis of Meynert, 6 and may predispose to hydroxynonenal toxicity and neural injury. 37 Moreover, GSTO1 has been reported to be involved in the activation of IL-1β, 13 a fundamental component in the inflammatory response, a mechanism that has been suggested as contributing to the pathogenesis of AD. 38,39 Indeed, several studies have suggested that IL-1β is overexpressed in the AD brain, 39 and is induced by the β-amyloid (Aβ) peptide. 40 The transcripts of GSTO1 were also significantly downregulated in the hippocampus of AD patients compared with that of controls. 14 In addition, GSTO1 can modulate the calcium release activity by ryanodine receptors present in the endoplasmic reticulum of many cells as well as in both presynaptic axon terminals and postsynaptic dendrites, where it regulates Ca2+-mediated processes involved in synaptic transmission and structural plasticity. 41 Aβ also directly induces oxidative stress in neurons through the production of reactive oxygen species (ROS), which may further disrupt Ca2+ homeostasis and exacerbate amyloidogenic processing of the amyloid precursor protein. A dysregulation of Ca2+ homeostasis may contribute also to tau protein hyperphosphorylation and self-aggregation, thereby promoting the formation of NFTs. 42 In this view, GSTO1 could interrupt a vicious neurodegenerative cycle, stopping intracellular increased Ca2+ concentrations.
Notwithstanding the present interesting findings, our study has several limitations. First, our sample size was relatively small for the demands of comprehensive analysis of haplotypes and stratified analysis with the APOE ε4 allele. However, our study had a statistical power of 85.8% and 98.3% for rs4925 C/A and A/A genotypes and of 99.0% for rs4925 A allele. It should be pointed out that our results might be influenced by several factors. Because AD is a complex disease in which environmental and genetic factors interact each other in the onset and development of pathophysiological mechanisms, the GSTO1 locus might have a small effect on the overall pathogenesis and clinical features of AD. Nevertheless, the significant association between AD and two haplotypes in GSTO1 gene cannot include the involvement of the locus as a whole in the pathogenesis and/or clinical expression of the disease, as suggested by the absence of important LD among the markers investigated. Moreover, interaction of this gene with additional other gene variants cannot be excluded by our findings. Thus, further studies evaluating rs4925 and rs1804834 and other GSTO1 gene variants on larger samples of AD patients with a different genetic background and combined metaanalysis across different studies are needed to clarify the role of GSTO1 locus in the pathogenesis of AD.
Footnotes
Acknowledgments
This work was supported by cofinanziamento MIUR 2003 (ex 60%), University of Foggia, and Ministero della Salute, IRCCS Research Program 2009-2011, Line 2: “Malattie complesse.” The funding agencies had no role in design or conduct of the study.