
Abstract
It has been suggested that increasing age is correlated with an acceleration of the progression of liver fibrosis induced by various agents, such as hepatitis C virus or chronic alcohol consumption. However, the cellular and molecular changes underlying this predisposition are not entirely understood. In the context of an aging population, it becomes challenging to decipher the mechanisms responsible for this higher susceptibility of older individuals to this acquired liver disorder. To address this issue, we induced liver fibrosis by carbon tetrachloride (CCl4) chronic administration to 8-week- and 15-month-old mice. We confirmed that susceptibility to fibrosis development increased with age and showed that aging did not affect fibrosis resolution capacity. We then focused on the impairment of hepatocyte proliferation, oxidative stress, and inflammation as potential mechanisms accelerating the development of fibrosis in the elderly. We detected no inhibition of hepatocyte proliferation after CCl4 injury in 15-month-old mice, whereas it was inhibited after a partial hepatectomy. Finally, we observed that, in a context in which liver oxidative stress was not differentially increased in both experimental groups, there was a higher recruitment of inflammatory cells, including mostly macrophages and lymphocytes, oriented toward a T helper 2 (TH2) response in older mice. Our data show that in conditions of equivalent levels of oxidative stress and maintained hepatocyte proliferative capacity, an increased inflammatory reaction mainly composed of CD4+ lymphocytes and macrophages expressing TH2 cytokines is the main factor involved in the higher susceptibility to fibrosis with increasing age.
Introduction
When hepatocytes are damaged by chronic injury, they release reactive oxygen species (ROS) and induce the recruitment of lymphocytes and macrophages. This leads to the activation of hepatic stellate cells, the major source of extracellular matrix deposition. Therefore, a vicious circle occurs in which inflammatory and fibrogenic cells stimulate each other and release ROS and cytokines. This eventually leads to an imbalanced process with more extracellular matrix produced that can be degraded. 10 Moreover, fibrosis may be precipitated by the premature and replicative hepatocyte senescence induced by ROS and continuous proliferation respectively. All of these parameters—fibrosis resolution, ROS production, inflammatory reaction, and hepatocyte proliferative capacity— are likely to be affected by age.
We investigated the reasons for the higher severity of liver fibrosis with age, by inducing chronic injury in the livers of 8-week and 15-month-old mice by carbon tetrachloride (CCl4) treatment. We confirmed that fibrosis levels were higher in older mice, despite the conservation of hepatocyte proliferative capacity, and demonstrated that the capacity to resolve fibrosis was not diminished by aging. We then studied oxidative stress and inflammation in aged and young chronically injured livers. An altered inflammatory reaction was identified as the major modification responsible for the higher level of liver fibrosis.
Materials and Methods
Ethics statement
All experiments were conducted in accordance with Institut National de la Santé et de la Recherche Médicale (INSERM) institutional guidelines for the care and use of laboratory animals. They were approved by the “Directeur départemental des Services Vétérinaires” of the Prefecture de Police de Paris (agreement number 75-1027).
Animal models
Two groups of male C57/BL6 mice purchased from Charles River were used in these experiments, one group of 8-week-old mice (n = 45) and one group of 15-month-old mice (n = 45). The animals had free access to food and water. Liver injury was induced by intraperitoneal injection of 3.5 mL/kg of CCl4 dissolved in a 1:10 (vol/vol) ratio with mineral oil. Control animals (Oil) received injections of the same volume of mineral oil. For acute studies, animals were injected once with CCl4 and killed after 36, 48, or 60 h, with bromodeoxyuridine (BrdU) (Sigma-Aldrich St Louis, MO) injected intraperitoneally 2 h before sacrifice at a concentration of 50 mg/kg (n = 6 animals per group and time point, n = 3 for oil-injected mice). For chronic studies, animals received two weekly intraperitoneal injections of CCl4 for 6 weeks and were killed 2, 7, or 14 days after the last injection (n = 6 animals per group and per time point). Blood was collected when the animals were killed, and serum alanine aminotransferase (ALAT) activity was measured on a Hitachi 747 analyzer. All experiments were conducted in accordance with institutional guidelines for the care and use of laboratory animals.
Histological analysis
Livers were harvested either in 10% buffered formalin and embedded in paraffin for histological evaluation. Liver sections (5 μm thick) were stained with Hematoxylin & Eosin (H&E) or with Picrosirius Red. For immunohistochemistry, sections were incubated with α-smooth muscle actin (α-SMA) antibody (M 0851, Dako, Glostrup, Denmark) or γH2AX antibody at a dilution of 1/50 dilution (#2577, Cell Signaling). For morphometric analysis, we studied 10 images per animal, from three different lobes at 100× magnification. The stained areas were quantified with ImageJ 1.37v software (NIH, Bethesda, MD). Ki67 and BrdU immunostainings were performed with a biotinylated monoclonal primary antibody (DAKO, Glostrup, Denmark) and developed with the Vectastain Elite ABC kit (Vector, Burlingame, USA) according to the manufacturer's instructions. For each liver sample, approximately 5,000–8,000 hepatocyte nuclei were counted on 15 fields per animal at 200× magnification. For assessment of inflammation, 5-μm-thick frozen sections were immunostained with CD68 antibody (MCA 1957; AbD Serotec, Oxford, UK), CD45 (clone 30-F11), CD4 (clone H129.19), and B220 antibodies (BD Pharmingen, San Diego, California) at 1/50 dilution as previously described in Strick-Marchand et al. 11
Real-time RT-PCR
RNA was purified from livers using the TRIzol method (Invitrogen). We then synthesized cDNA with the Transcriptor First Strand cDNA synthesis kit (Roche, Mannheim, Germany). Quantitative reverse transcriptase polymerase chain reaction (RT-PCR) was performed in duplicate using the QuantiTect SYBR Green PCR kit (Qiagen, Mainz, Germany) in a LightCycler apparatus (Roche). QuantiTect Primer Assays (Qiagen) were used to amplify all of the genes analyzed with the standard QuantiTect protocol. Relative expression levels were calculated and normalized with respect to the control gene, hypoxanthine-guanine phosphoribosyl transferase (HPRT). Wild-type mice receiving injections of excipient (Oil) were used as a reference for calculating the fold-induction of gene expression.
Mitochondrial function and oxidative stress analysis
Liver homogenates were resuspended in 0.1 mM phosphate buffer. Complex I and citrate synthase activities were measured as previously described. 12 For the evaluation of hepatic lipid peroxidation, we measured thiobarbituric acid reactants (TBARs) as previously described. 12 TBARs levels are expressed in nanomoles of malondialdehyde (MDA) equivalents per milligram of protein.
The activities of the antioxidant enzymes catalase, glutathione peroxidase (GPx), and Mn and CuZn superoxide dismutase (SOD) were determined as previously described 13 using concentrations of the tissue extracts in which the assays were linear and expressed as relative units per milligram of protein. We evaluated 8-hydroxy-2′-deoxyguanosine (8-oxo-dG) immunofluorescence according to the kit manufacturer's instructions for paraffin-embedded tissues. Briefly, sections were incubated with 100 μg/mL RNase A, 150 mM NaCl, and 15 mM sodium citrate for 1 h at 37°C. DNA was then denatured by immersion in 2 M HCl for 5 min and neutralized in 1 M Tris base. After blocking with 10% fetal bovine serum, tissues were stained with anti-8-oxo-dG (Trevigen, Catalog No. 4354-MC-050, Clone 2E2) at 1:300 dilution overnight at 4°C. Anti-8-oxo-dG binding was detected using a Alexa Fluor 488 antibody with counterstaining of nuclei with Hoechst.
Western blotting
Total protein extracts were obtained from snap-frozen tissue by homogenization in lysis buffer as previously described. 14 Immunoblotting was performed with anti-metalloproteinase-2 (MMP-2) and anti-cyclin D1 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). A γ-tubulin-GTU-88 antibody (Sigma, St. Louis, MO) was used as an internal control. Enhanced chemiluminescence was developed with a horseradish peroxidase (HRP)-conjugated secondary anti-mouse antibody (Dako).
Statistical analysis
Statistical analyses were performed with Statview 4.5 software. The Student t-test was used for comparisons. Data are presented as mean ± standard error of the mean (SEM). p values below 0.05 were considered significant.
Results
Increase in CCl4-induced fibrosis with age
CCl4 induces centrilobular necrosis with the recruitment of inflammatory cells to the injured area. When repeatedly injected, it is responsible for the activation of hepatic stellate cells resulting in the development of fibrosis. We injected CCl4 twice a week into 15-month- and 8-week-old male mice during 6 weeks and assessed serum ALAT levels, reflecting cytolysis, and histological signs of liver necrosis, 2 days after the last injection. Our results showed that younger mice displayed significantly lower levels of liver injury (Fig. 1A,B).

Increased liver necrosis with age after chronic liver injury. (
We then quantified liver fibrosis by Picrosirius Red staining 2 days after the last CCl4 injection, a time point known to correspond to the peak of fibrosis. We found that the fibrosis level was significantly higher in 15-month-old animals than in 8-week-old mice (Fig. 2A). We then analyzed the activation of fibrogenic cells by determining alpha-SMA levels, which were found at 2.5-fold times higher levels in older animals (Fig. 2B). The higher levels of mRNA expression for early markers of fibrogenesis, such as transforming growth factor-β1 (TGF-β1) and collagen-α1, confirmed these findings (Fig. 2C). These results demonstrate that aging contributes to the progression of liver fibrosis.
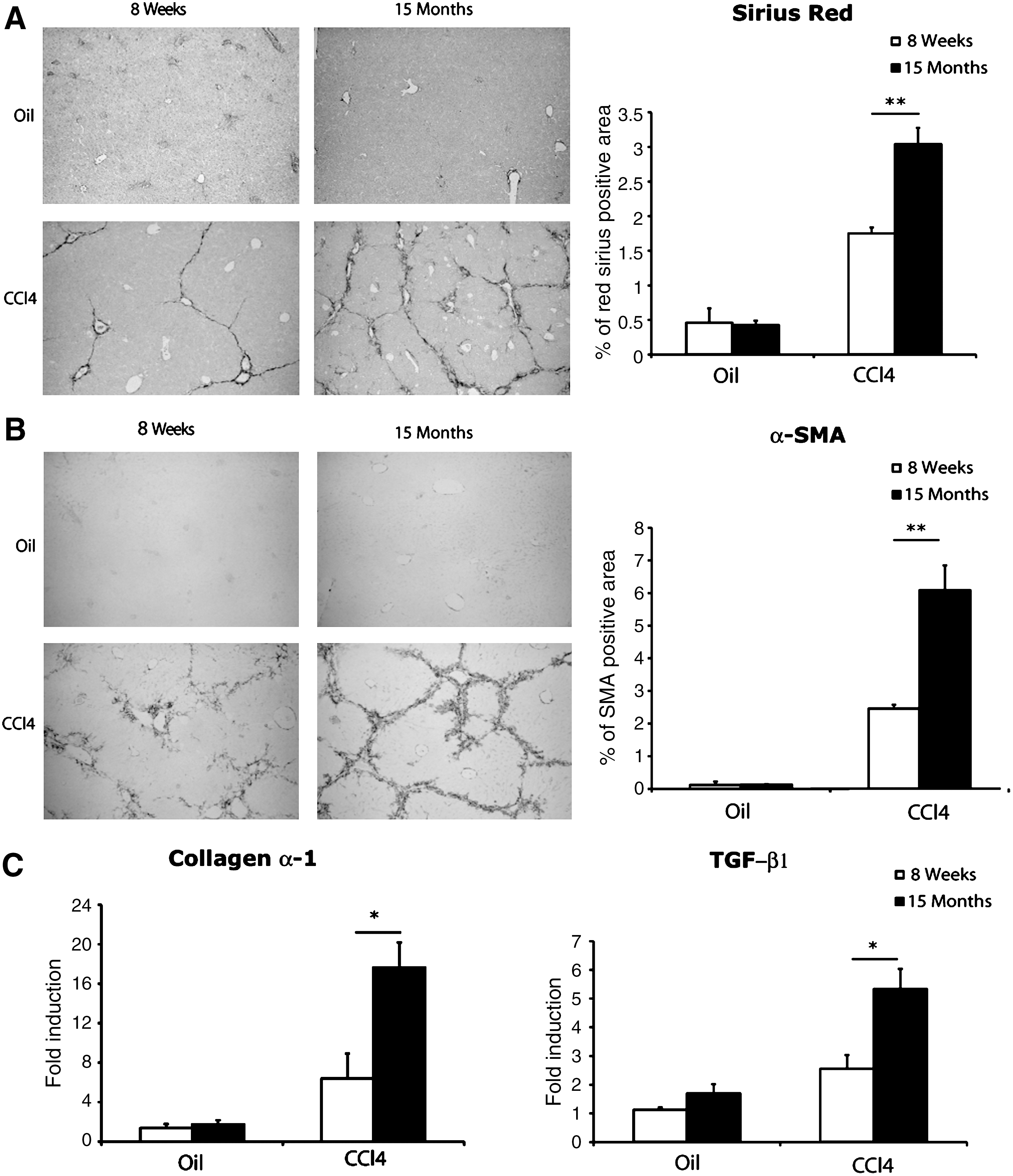
Increased susceptibility to liver fibrosis with age. (
Aging does not affect the resolution of fibrosis
Liver fibrosis is the result of an imbalance between the continuous processes of extracellular matrix production and degradation. Between each toxic injection, fibrosis spontaneously regresses by fibrolysis. We hypothesized that an impairment of fibrolysis in older animals might be responsible for the higher level of fibrosis. We analyzed the consequences of aging on fibrosis resolution, by treating animals for 6 weeks with CCl4 as in the above experiment, but killing them 2, 7, or 14 days after the last injection of CCl4. After 7 days of spontaneous recovery, collagen deposition was found to be decreased by 56% in young mice and 59% in aged mice (Fig. 3A,B). By day 14, the difference in fibrosis levels between 8-week and 15-month animals was no longer significant. These data indicate that increasing age does not hamper the capacity of the liver to resolve fibrosis at least at 15 months of age. Resolution of fibrosis involves the activation of various MMPs, mirrored by a decrease in the level of tissue inhibitors of metalloproteinases (TIMPs) expression.
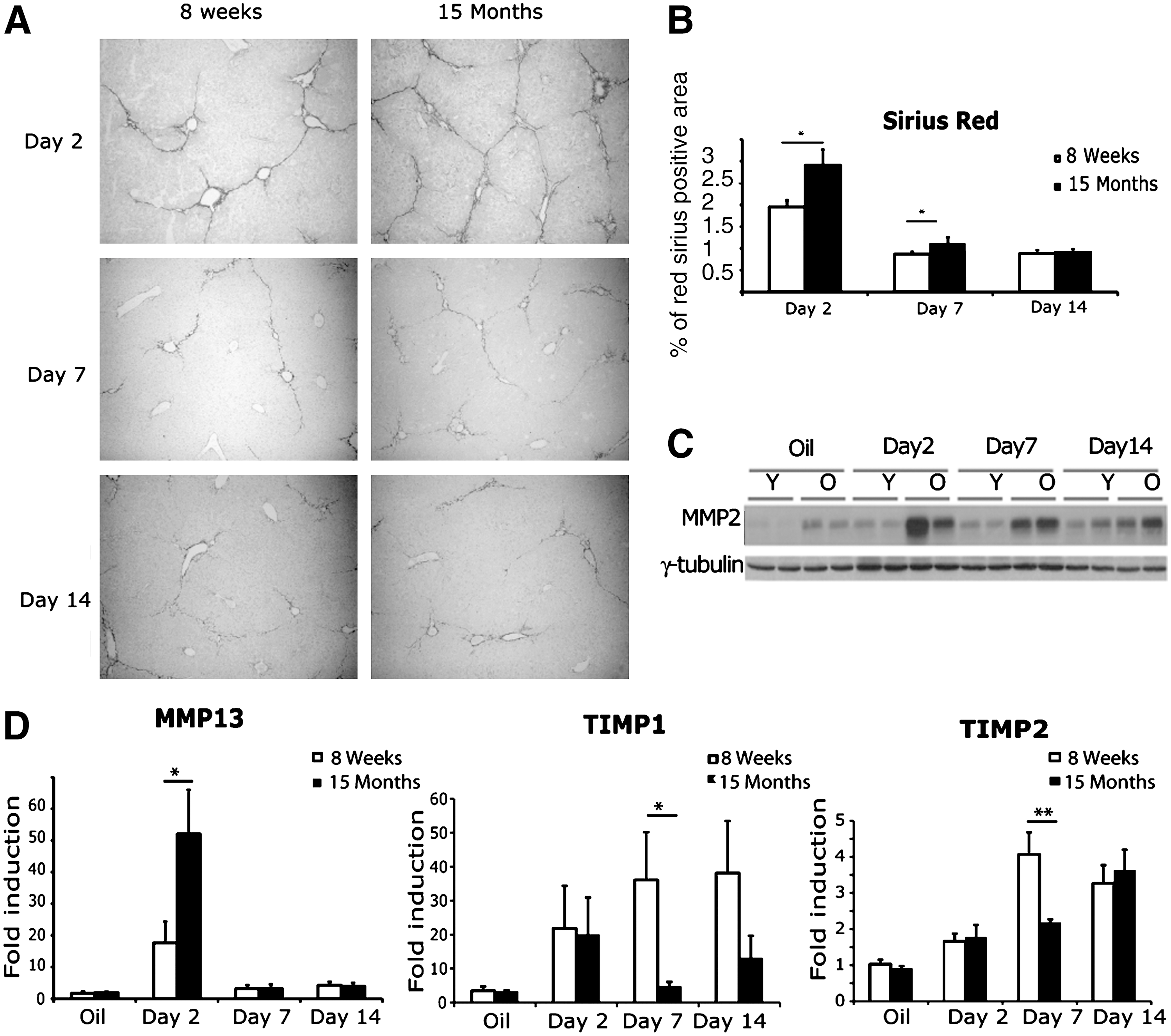
Aging does not hamper the resolution of liver fibrosis. (
Thus, we looked at MMP-2, MMP-13, TIMP-1, and TIMP-2 expression at 2, 7, and 14 days after the last CCl4 injection. MMP-2 protein levels were significantly higher in older mice before treatment. These levels increased in both groups 2 and 7 days after the end of the chronic injections, and remained significantly higher in 15-month-old mice than in 8-week-old mice at these time points (Fig. 3C). The difference was no longer significant between the two groups by day 14. MMP-13 mRNA levels were also higher in the older group of mice 2 days after the last injection (Fig. 3D). TIMP-1 and TIMP-2 mRNA levels were much lower in 15-month-old mice than in 8-week young mice 7 days after the last CCl4 injection (Fig. 3D). These expression profiles indicate that older livers produce more MMP-2 and MMP-13 in correlation with less TIMP-1 and TIMP-2 during the resolution phase, resulting in the degradation of extracellular matrix and the efficient resolution of fibrosis.
Aging does not reduce the capacity of mouse hepatocytes to proliferate after CCl4 injury
One of the most important age-related changes in liver function is a decrease in its regenerative capacity. 15 This reduced hepatocyte proliferation with age has been studied mostly in vivo after two thirds partial hepatectomy (2/3PH). 16 An impaired hepatocyte proliferation induced by forced telomere shortening has been associated with the accelerated development of liver cirrhosis in response to chronic injury in mice. 17 Therefore, we hypothesized that an impairment of hepatocyte proliferation capacity with age could have participated in the higher levels of liver fibrosis. Surprisingly, the proportion of ki67-positive hepatocytes was equivalent in young and old mice at the end of CCl4 treatment (Fig. 4A). Moreover, we observed hepatocytes in various phases of mitosis in the livers of both groups, indicating that 15-month-old hepatocytes were able to complete the cell cycle in vivo after CCl4 injury (Fig. 4A). In contrast, 15-month-old mice showed a drastic impairment of hepatocyte proliferation 36 hr after PH, a timing that corresponds to the expected peak of hepatocyte DNA synthesis in 8-week-old animals (Fig. 4B). Moreover, no mitotic figures 48 hr after PH were observed in aged animals (data not shown).
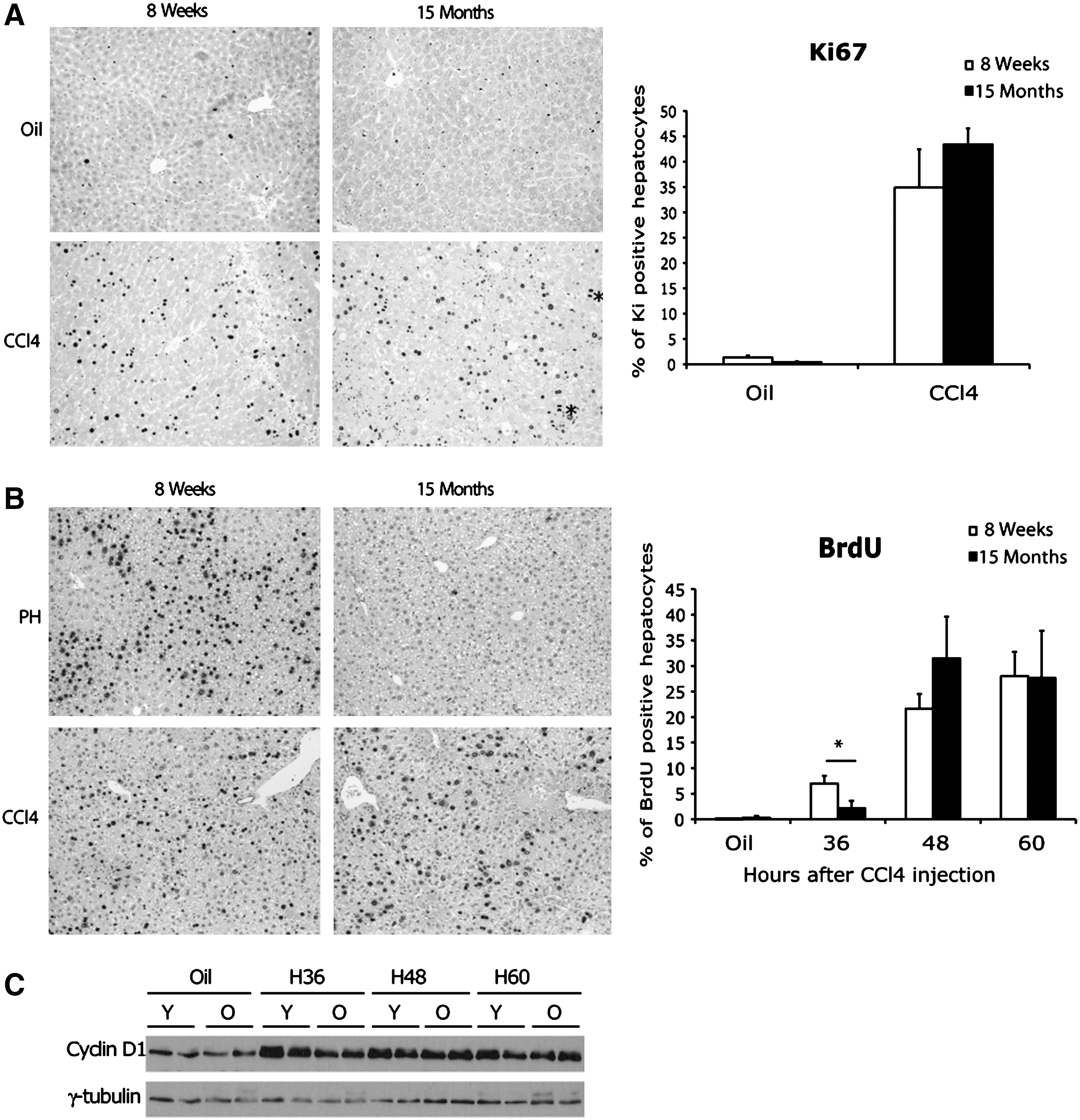
Fifteen-month-old hepatocytes show an impaired proliferative capacity after partial hepatectomy but not after carbon tetrachloride (CCl4) injury. (
We wondered whether this unexpected maintained proliferative capacity of aged hepatocytes after toxic injury might be due to the progressive selection, by repeated CCl4 administration, of a highly proliferative population of hepatocytes. We tested this hypothesis by carrying out another set of experiments in which we treated young and aged mice with a single dose of CCl4 and analyzed BrdU staining at various time points. Equivalent DNA synthesis peaks were observed in 8-week- and 15-month-old mice at 48 and 60 hr after CCl4 injection (Fig. 4B). These findings were confirmed by an analysis of cyclin D1 expression, normally induced during G1/S transition phase, which displayed similar patterns of expression in both groups of mice after CCl4 injury (Fig. 4C). Thus, in our model, the higher level of fibrosis in 15-month-old mice was not due to an impaired capacity of their hepatocytes to enter the cell cycle. However, an analysis of γ-H2AX staining, which reveals DNA damage at either uncapped telomeres or persistent DNA strand breaks, 18 showed that the percentage of stained hepatocytes was higher in older livers, suggesting that the livers of 15-month-old mice were more susceptible to DNA breaks than young ones (Supplementary Fig.1).
CCl4-induced oxidative stress is equivalent in 8-week and 15-month-old mice
We evaluated the global oxidative stress induced by CCl4 administration during the progression of fibrosis. We first quantified lipid peroxidation (malondialdehyde) in liver homogenates. Chronic CCl4 injection induced lipid peroxidation in both groups, with no significant difference between young and aged mice (Fig. 5A). We then used 8-OHdG as a reliable marker of oxidative DNA damage that stains cell nuclei. Only a small proportion of hepatocyte nuclei were free of DNA damage, but this proportion was similar in aged and young injured livers (Fig. 5D). We then investigated the specific effects of an increased ROS production on mitochondrial respiratory chain activity and found that complex I activity was significantly decreased by CCl4 injury, with no significant difference between aged and young mice (Fig. 5B). Similar results were obtained for complex IV (data not shown). We then investigated defense systems that protect against ROS in the livers of young and aged mice by quantifying the activity of Mn-SOD, CuZn-SOD, GPx, and catalase in the livers of these animals. Liver injury significantly decreased the activities of all these enzymes. However, activity levels were similar in old and in young injured livers for all of the enzymes tested except catalase (Fig. 5C). Altogether, these findings indicate that there was no overall higher level of mitochondrial or extramitochondrial liver ROS production in 15-month-old mice compared to 8-week-old mice after chronic injury.

Oxidative stress induced by chronic carbon tetrachloride (CCl4) liver injury is equivalent in young and aged mice, assessed by malondialdehyde (MDA) levels (
Alteration of the inflammatory reaction with age after CCl4 liver injury
Both macrophage and lymphocyte compartments have been shown to deteriorate progressively with advancing age. 19 However, they have not been studied after chronic liver injury. Using ImageJ software analysis, we quantified the areas of inflammation 2 days after the last CCl4 treatment and found a significantly higher level of inflammatory cell recruitment in 15-month-old than in 8-week-old livers (Fig. 6A). We found by immunostaining that scar-associated macrophages (CD68) and leukocytes (CD45), among which mainly CD4+ cells, were more numerous in the fibrotic areas with increasing age (Fig. 6B). By contrast, no clear difference was observed for B lymphocytes (Fig. 6B), or CD8-positive cells (data not shown). We assessed the global activity levels of these cells, by analyzing the liver expression profiles of various cytokines by real-time RT-PCR after chronic liver injury. We observed a complex modulation of cytokine expression profile with age, but without significantly higher levels of induction for macrophage inflammatory protein-1 (MIP-1α), tumor necrosis factor-α (TNF-α), monocyte chemotactic protein-1 (MCP-1), or interleukin-6 (IL-6). Interferon-γ–inducible protein (IP10) was the only cytokine for which significantly higher levels of induction were observed in 15-month-old than in 8-week-old mice. All of these proinflammatory cytokines correspond to a T helper 1 (TH1) expression profile. Studies in various cytokine-deficient mice have shown that fibrogenesis is linked with the development of a T helper 2 (TH2) CD4+ T cell response. 20
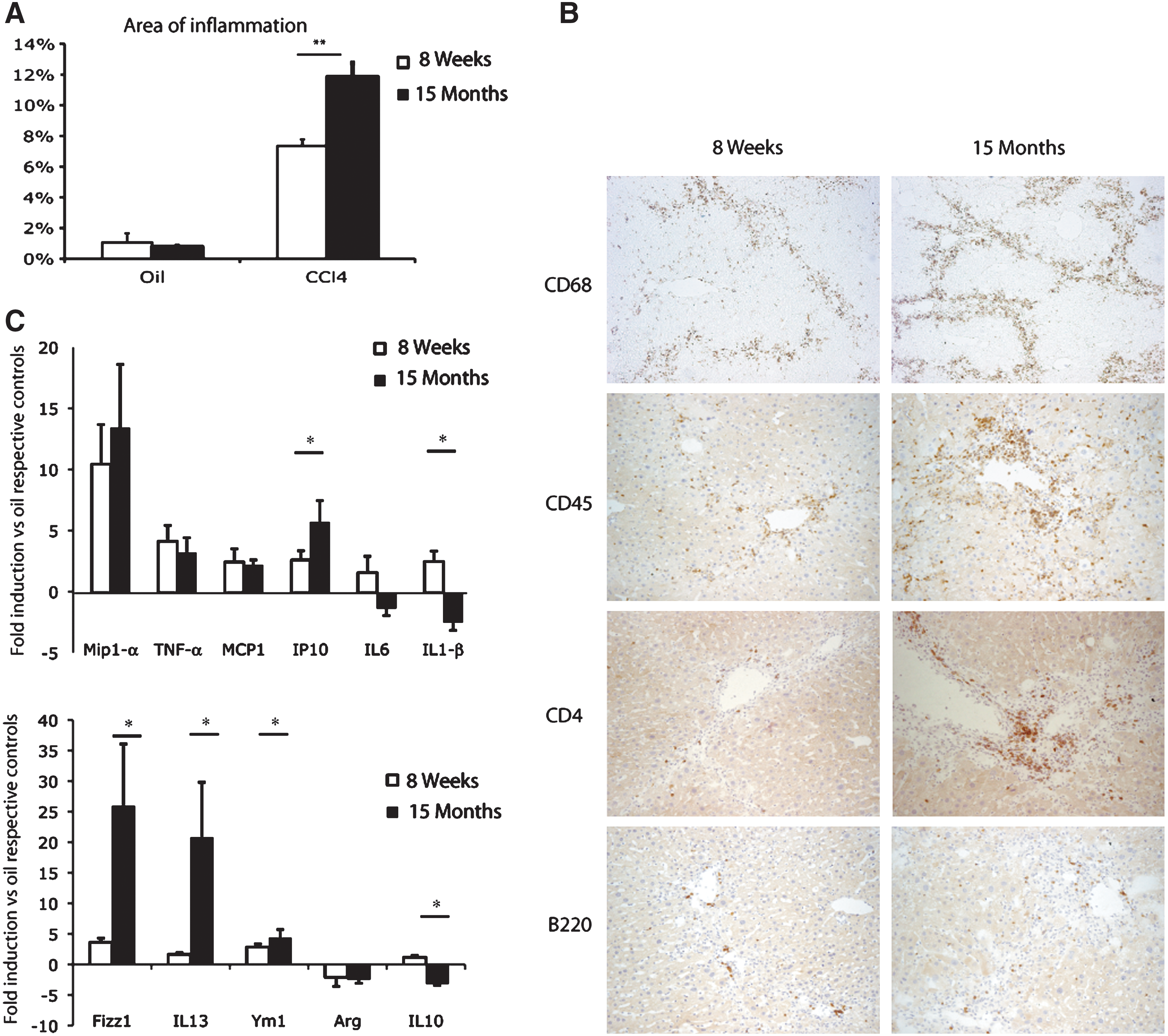
Modulation of the inflammatory reaction in the injured liver of 8-week-old and 15-month-old mice. (
Therefore, we analyzed the expression of several genes associated with the TH2 response. We observed significantly stronger induction of IL-13, Fizz1, and Ym1 in the livers of 15-month-old mice compared to young mice, suggesting a global increase in the strength of induction of the TH2 response with age after CCl4 injury (Fig. 6C). Arginase mRNA expression was downregulated both in young and old mice. However, the relative mRNA expression was significantly lower in 15-month-old mice compared to young ones (data not shown). Finally, whereas 8-week-old livers displayed weak but significant induction of IL-10 with chronic aggression, 15-month-old livers showed a significant decrease in the levels of this cytokine (Fig. 6C). These data highlight the existence of a complex age-associated dysfunction of the inflammatory response to chronic toxic injury, with an increase in alternative macrophage activation.
Discussion
In this study, we induced liver fibrosis by chronic administration of CCl4 in two groups of animals differing in age. CCl4 hepatotoxicity results from its metabolism in a toxic trichloromethyl free radical by CYP2E1. The conversion of CCl4 into this toxic compound has already been shown to occur at similar rates in young and aged rodents after the intraperitoneal administration of CCl4, and no significant difference in Cyp2E1 activity between the age groups has been observed. 21 –23 Similarly, we found no difference in serum ALAT levels between young and old animals at different time points after a single CCl4 injection (data not shown). After 6 weeks of chronic liver injury, we found a higher level of necrosis in old mice as shown by H&E staining and by the determination of serum ALAT levels. This greater degree of liver injury at the end of the treatment was correlated with the higher level of fibrosis in aged mice.
Fibrosis occurs when more extracellular matrix is produced than degraded. Macrophages play a key role in matrix degradation particularly through the secretion of MMPs. 24,25 Increase in MMP-2 and MMP-13 levels together with a decrease in TIMP-1 and TIMP-2 levels have been shown to facilitate the rapid resolution of experimental liver fibrosis. 26 –28 We found that MMP-2 and MMP-13 were overproduced in old mice 7 days after the last injection. TIMP-1 and TIMP-2 levels mirrored those of MMP-2 and MMP-13, with significantly lower induction observed in aged than in young mice. This expression profile led to the regression of fibrosis even more rapidly in old livers than in young livers. These results confirm that the resolution of fibrosis was not impaired with aging.
The hepatocytes of the adult liver are quiescent cells with a remarkable capacity to re-enter the cell cycle, enabling the liver to regenerate after injury. Many studies have demonstrated an impairment of the regenerative capacity of the liver in old rodents after 2/3 PH. These publications reported a much smaller DNA synthesis peak in aged rodents, consistent with impaired S phase entry in the hepatocytes of old animals. 16 We also found an impaired hepatocyte proliferative capacity in 15-month-old mice compared to 8-week-old mice after PH. In contrast, we observed no such decrease in the proportion of proliferating hepatocytes after either repeated or acute CCl4 injections in 15-month-old mice. Interestingly, it has also been demonstrated that aged hepatocytes retain their replicative capacity in response to a mitogen. 29
Therefore, our data suggest that liver regeneration in response to toxic aggression by CCl4 involves mechanisms distinct from those observed in the PH model to inducing hepatocytes S phase entry, an idea that has been recently documented with lineage strategies. 30 Mice have constitutively longer telomeres than humans, which could minimize any cell cycle blockade due to telomere shortening after chronic aggression. We cannot exclude the hypothesis that replicative senescence, as observed in human fibrotic liver samples, 31 contributes to the increase in fibrosis with age observed in humans.
Oxidative stress plays a major role in the pathogenic development of liver fibrosis, regardless of disease etiology. Oxidative damage is also known to accumulate with age, particularly in the brain, and aging is generally correlated with a reduced tolerance to oxidative stress (for review, see ref. 32). Conflicting results have been reported for the liver. Hepatocytes isolated from aged rats have been shown to be more sensitive to oxidative stress in vitro. 33 However, no increase in ROS production has been observed in the livers of old mice and mitochondrial DNA alterations have not been observed before the age of 19 months. 34
The age-associated modulation of major antioxidant proteins in the quiescent liver has also been the subject of controversy. 35 –38 However, none of these markers has been studied after chronic injury. In our model, despite the induction of lipid peroxidation and mitochondrial dysfunction together with a decrease in the activity of antioxidant enzymes, we detected no difference in the levels of these oxidative stress markers between young and old mice after injury except for catalase activity. Therefore, the higher susceptibility to fibrosis development in old mice in our model cannot be explained by an increase in mitochondrial dysfunction and global oxidative stress with age. However, our results suggest a decreased tolerance to the major oxidative stress induced by CCl4 with age. This DNA damage, if not correctly repaired, could indeed cause the progressive failure of the cellular machinery, accelerating the progression of liver fibrosis with age. Further studies are required to determine whether this accumulation of DNA breaks will ultimately leads to abnormal cell division.
Liver inflammation is the hallmark of early-stage liver fibrosis, leading to hepatic stellate cell activation and extracellular matrix deposition. The recruitment of macrophages 25,14 and lymphocytes 39 plays an essential role in the progression of this disease. We identified a significantly higher level of macrophages and CD4+ cells recruitment in the liver of 15-month-old mice than of 8-week-old mice after injury. Macrophage activation was initially described as a TH1-interferon-γ (IFNγ) -mediated process. We analyzed the chemokine and cytokine expression profile of these inflammatory cells after 6 weeks of CCl4 treatment and found a complex pattern of differential expression with age. MIP-1α and MCP-1, two chemokines identified as essential profibrotic mediators, were induced to similar extents in old and young mice. Similar levels of induction were also observed for TNF-α and IL-6. IL-1 has recently been shown to be important in both early fibrogenesis and the late phase of fibrosis maintenance. 40 By contrast to the increase in IL-1β levels observed in young livers, downregulation of this cytokine was detected in aged livers. Finally, of all the TH1 cytokines tested, IP10 (IP-10/CXCL10), for which intrahepatic mRNA levels were correlated with both inflammation and fibrosis in hepatitis C patients, 41 was the only protein produced in larger amounts in old mice than in young ones.
Macrophages differentiate into at least two functionally distinct populations, depending on whether they are exposed to TH1 or TH2 cytokines. Many of the genes associated with TH2 responses and alternative macrophage activation have been implicated in the mechanisms of wound healing and fibrosis (for review, see ref. 20). Most of the TH2-dependent cytokines tested in our animals were indeed found up-regulated in old fibrotic livers compared to young ones. In correlation with a higher recruitment of CD4 lymphocytes, we observed a significant up-regulation of IL-13, and of IL-13–induced genes such as Fizz-1 (Found in Inflammatory Zone 1) and to a lesser extent, Ym1. IL-13 is produced mainly by CD4+TH2 cells and has been described as the main profibrotic mediator among TH2 cytokines in the liver 42 and to be correlated with the fibrotic stage in patients with HCV infection. 43 Moreover, Fizz-1 has recently been shown to induce myofibroblast differentiation in pulmonary fibrosis through α-SMA expression and Notch1 signaling. 44 IL-10 and arginase-1 showed different modulations of their expression profile, being downregulated in old injured livers. Interestingly, they are both considered to be immunosuppressive cytokines that downregulate chronic inflammatory responses and reduces hepatic fibrosis in mice. 45 –47 Reduced IL-10 and arginase-1 expression has been recently correlated with a higher level of fibrosis in CXCR3-deficient CCl4-injured livers. 48 Altogether, the dysregulated inflammatory reaction observed in old mice may thus account for the higher level of fibrosis in these animals.
In conclusion, using a mouse model of toxic-induced chronic liver injury, we confirmed that the susceptibility to liver fibrosis increases with age. Our data provide further evidence for the higher recruitment of inflammatory cells and the major role of the altered inflammatory reaction associated with aging in this process even in the absence of a higher oxidative stress level. This inflammatory reaction is oriented principally toward alternative macrophage activation, with an increase in TH2 cytokine production. Our data therefore help to better understand the mechanisms behind the higher susceptibility to fibrosis with age.
Footnotes
Acknowledgments
We thank Drs. H Strick-Marchand, C. Desdouets, and B. Chazaud for fruitful discussions. We also thank Dr. P. Jaffray for ALAT determinations. This study was supported by the Agence Nationale de la Recherche (ANR) and Institut National du Cancer (INCA).
Author Disclosure Statement
The authors have no financial conflicts of interest to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.