
Abstract

Damage to the mitochondria has been hypothesized to play a critical role in cellular and organismal aging. Mitochondrial theories of aging hypothesize that reactive oxygen species (ROS) generated during aerobic respiration damage proteins, lipids and DNA in the mitochondria.
1
Accumulated damage to the respiratory chain increases the generation of ROS, which contributes to mitochondrial dysfunction either directly, through faulty mitochondrial DNA (mtDNA)-encoded proteins,
2
or via the lysosomal–mitochondrial axis
3
in which damage to both organelles combines to reduce cell function. Mitochondrial homeostasis has been incorporated into these models, most often emphasizing how ROS-mediated damage attenuates autophagy or biogenesis, thus leading to cellular dysfunction. Loss of telomere length in somatic cells presents a potential barrier to life span and health span enhancement. Most somatic mammalian cells lack telomerase activity, which results in a limited replicative life span called the “Hayflick limit,” named after its discoverer Leonard Hayflick.
4
Telomeres shorten with each cell division until the cell senses the short telomeres as DNA damage, stops dividing, and enters a “replicative senescent” state. The absence of telomerase in somatic cells has been hypothesized to be an anticancer adaptation,
5
because it must be overcome for a transformed cell to form a clinically relevant tumor. Although, the connection between short telomeres and normal human aging is yet to be established conclusively, the absence of telomerase in most somatic cells presents a theoretical barrier to strategies for engineered negligible senescence (SENS) that would have to be overcome at least transiently.
6
The Hayflick limit and mitochondrial dysfunction have largely been seen to be unconnected until now.
Sahin and colleagues 7 report a fascinating study linking telomere and mitochondrial dysfunction. So-called “G4” transgenic mice accumulated short telomeres over four generations due to the absence of either the telomerase reverse transcriptase (Tert) or telomerase RNA component (Terc) genes. As expected, the mice accumulated pathologies associated with proliferating cells (“stem cell depletion”). However, they also exhibited cardiomyopathy and liver dysfunction (glucose intolerance and reduced capacity for detoxification), pathologies in organs with predominately nonproliferating cells. Hematopoietic stem cells (HSCs), liver cells, and heart cells from G4 mice had reduced mitochondrial function, including reduced adenosine triphosphate (ATP) synthesis, fewer mitochondria, reduced levels of some ROS detoxifying enzymes, and increased carbonylated proteins and ROS levels. Together, these findings indicate a profound level of mitochondrial dysfunction similar to that seen in old animals or humans.
Messenger RNA (mRNA) profiling revealed that master transcriptional regulators, peroxisome proliferator-activated receptor-γ (PPAR-γ), co-activator 1 α and β (PGC-1α and PGC-1β) were significantly reduced. These genes play a critical role in mitochondrial biogenesis (see ref. 12, below). G4 mice bearing reduced levels of PGC-1α/β exhibit impaired gluconeogenesis, cardiomyopathy, and reduced ability of HSCs to reconstitute bone marrow. Adenoviral expression of the PGC-1α gene in the liver of G4 mice restored mitochondrial function and gluconeogenesis. The authors hypothesize that activation of p53 due to the presence of dysfunctional telomeres is responsible for the repression of PGC-1α and PGC-1β. To support this contention, they found that p53 binds the PGC-1α and PGC-1β promoters and conversely that G4 mice lacking p53 exhibit substantial PGC-1α and PGC-1β expression. In these mice (p53 −/− × Terc −/−), reduction of p53 partially rescues the mitochondrial, heart failure, and gluconeogenesis phenotypes that result from the short telomeres. Taken together these data strongly suggest that short telomeres, and perhaps other DNA damage or even other types of cell stress can cause p53-mediated suppression of PGC-1α and PGC-1β, which, in turn, leads to mitochondrial dysfunction similar to that seen in normal aging.
It would be interesting to test whether downregulation of PGC-1α or PGC-1β can partially explain the shorter life span seen in transgenic mice that express hyperactive p53. 8,9 Mitochondrial dysfunction resulting from repression of PGC-1α and PGC-1β is not unexpected because these master regulators of mitochondrial biogenesis are necessary for mitochondrial homeostasis and are known to be expressed at lower levels in old animals. These proteins are positioned at a signaling nexus that integrates cellular response to ROS and mitochondrial metabolism. Given the strong phenotypes seen in this paper, we hypothesize that differential regulation of mitochondrial biogenesis via PGC-1α and PGC-1β signaling may play a more primary role than previously thought in increasing mitochondrial dysfunction with age and represent a good target for health span enhancement. In turn, signaling from the mitochondria may play a more fundamental role in aging than previously assumed as well. For example, Durieux et al. 10 recently showed that electron transport chain (ETC)-mediated longevity in Caenorhabditis elegans is communicated to distal cells by a mitochondrial stress-triggered unfolded protein response (UPR) by an unidentified mitokine.
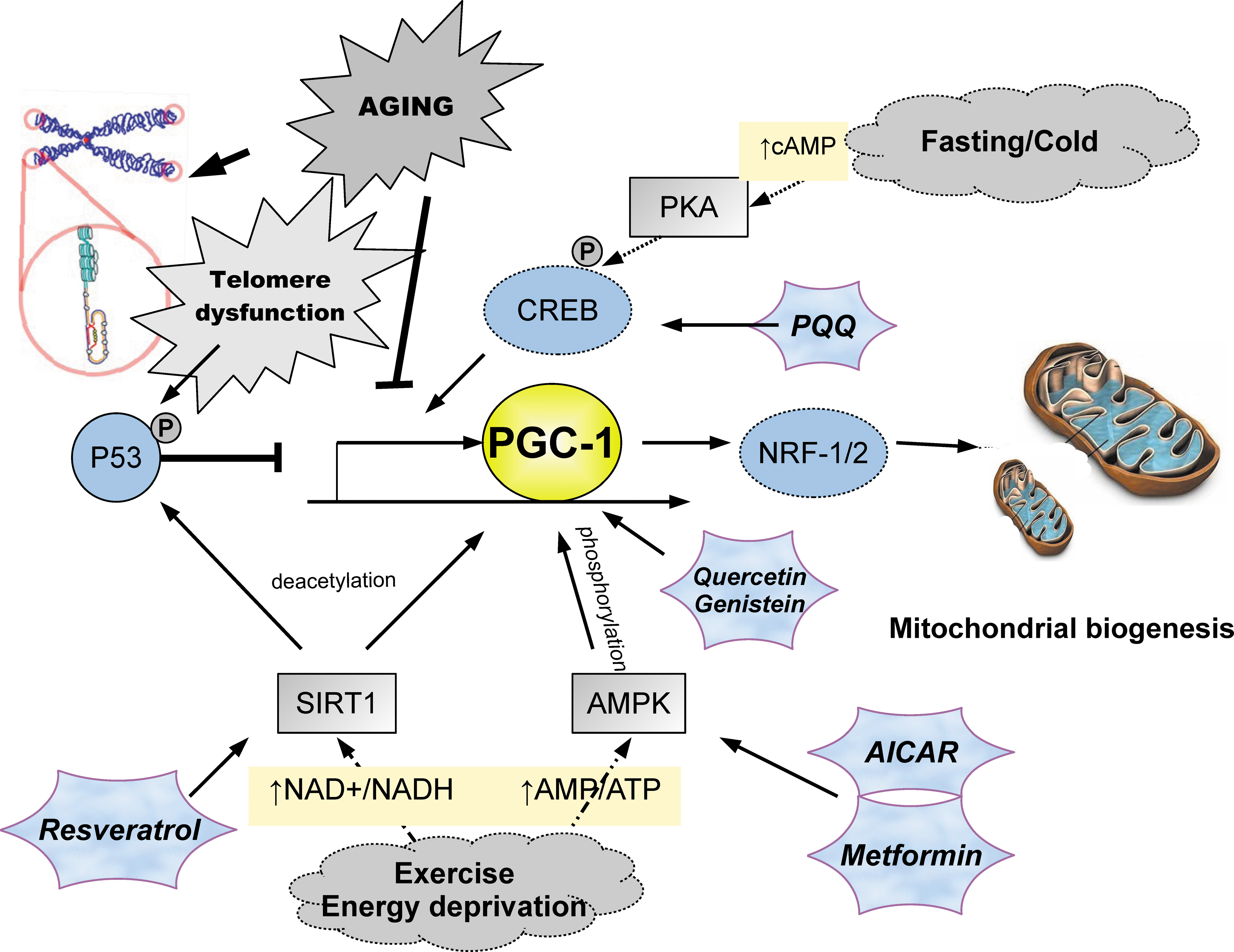
Peroxisome proliferator-activated receptor-γ co-activator 1 (PGC-1) signaling pathways control mitochondrial biogenesis. Cellular stresses associated with aging, including telomere dysfunction, dietary excess and inactivity down-regulate activity of master regulators PGC-1α/β through p53. Exercise, fasting, and exposure to cold upregulate PGC-1 through stimulation of sirtuin 1 (SIRT1), adenosine 5′-monophosphate-activated protein kinase AMPK, and cyclic AMP response element-binding (CREB). Active PGC-1 stimulates mitochondrial biogenesis through increased NRF1/2. Several promising pharmaceutical interventions shown to modulate PGC-1 are shown (hexagonal stars). (Color image is available at
Medical Implications of This Work
The work of Sahin et al. 7 suggests that p53, telomerase, and/or PGC-1α and PGC-1β may be interesting targets for therapeutic intervention for aging-associated diseases. However, the association of p53 inactivation or telomerase reactivation with potential tumorigenesis should inspire great caution. Therefore, we focus below on PGC-1α and PGC-1β. That forced expression of moderate amounts of PGC-1α in skeletal muscle protects aging mice from sarcopenia, loss of bone mineral density, increased inflammation, and loss of insulin sensitivity 11 is further reason for exploring these targets.
Regulation of PGC-1α and PGC-1β
What do we know? Many reports support the role of PGC-1α and PGC-1β as master regulators of mitochondrial biogenesis. Overexpression of PGC-1α or PGC-1β in cultured cells or in transgenic mice results in massive increases in mitochondrial content. Knockout of both PGC-1α or PGC-1β leads to death of newborn mice within a few days, and these mice have fewer mitochondria. 12 Skeletal muscle is significantly affected by overexpression of these proteins: PGC-1α inhibits muscle atrophy in animals that do not exercise 13 and confers strongly enhanced exercise capability on such mice, 14 and PGC-1β improves insulin resistance in skeletal muscle. 15 Thus, it is likely that stimulation of PGC-1α and PGC-1β will benefit health span.
Molecular mechanisms of action
PGC-1α has a potent amino-terminal transcriptional activation domain, whereby it interacts with chromatin remodeling co-activator complexes, including GCN5, SRC-1, and CBP/p300. It also binds the promoter regions of many transcription factors and nuclear hormone receptors including PPAR-γ, ERRa, which controls fatty acid oxidation, and NRF-1, YY1, PPAR-α, and MEF2C, which act on the respiratory chain. PGC-1β lacks the RNA-processing domain of PGC-1α, but retains much of the same ability to stimulate multiple nuclear respiratory genes. PGC-1β promotes a much higher level of coupled respiration than PGC-1α because of differences in proton leakage. 12 PGC-1α and PGC-1β stimulate mitochondrial biogenesis by stimulating expression of these downstream targets.
PGC-1's are regulated by Sirtuin 1 and adenosine5′- monophosphate–activated kinase
PGC-1α is activated by the nicotinamide adenine dinucleotide (NAD)-dependent deacetylase sirtuin 1 (SIRT1). The SIRT1 homolog SIR2 is associated with increased replicative life span in yeast and increased longevity in C. elegans. 16 SIRT1 deacetylates multiple lysine residues on PGC-1α, promoting mitochondrial fatty acid oxidation in response to low glucose. Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK), a sensor that is activated upon energy depletion in muscle and is known to stimulate mitochondrial biogenesis as well, 17 phosphorylates PGC-1α on threonine-177 and serine-538. AMPK mediates at least some of its effects to stimulate mitochondrial biogenesis via PGC-1α. AMPK is downregulated during aging. 18 SIRT and AMPK pathways cooperate to mediate calcium-dependent mitochondrial proliferation in myocytes. Therefore, stimulation of SIRT1 and AMPK should help activate PGC-1 proteins.
Current availability of drugs or activities that stimulate PGC-1α or PGC-1β
Given the potential benefits of augmented PGC-1α and PGC-1β function, new drugs to increase expression or activity of these proteins are expected to have clinical benefit. Several existent drugs or nutriceuticals known to increase the activity of AMPK or SIRT1 also stimulate PGC-1 proteins, at least indirectly. For example, metformin is known to stimulate AMPK via inhibition of AMP deaminase, 19 and multiple studies indicate stimulation of PGC-1. 20,21 Resveratrol, which stimulates SIRT1, possibly indirectly, 22 and indirectly stimulates AMPK, upregulates PGC-1α activity. 23,24 Dietary supplementation with pyrroloquinoline quinone (PQQ), a redox cofactor, increases mitochondrial biogenesis and function in mice. 25,26 In these mice, PGC-1α is elevated and required for PQQ effects. PGC-1α is stimulated through increased phosphorylation of cyclic (c) AMP response element-binding (CREB) on Ser-133, which itself is required for PGC-1α upregulation. 26 Additional compounds that stimulate PGC-1α include genistein and other isoflavones 27 and quercetin. 28 Finally, AICAR (5-aminoimidazole-4-carboxamide-1-b-D-ribofuranoside), an experimental drug that increases AMPK activity, also stimulates PGC-1α. 24,29
Moderate exercise may be the safest means to increase PGC-1. Elevated PGC-1 protein expression has been reported in exercised animals and humans, with P38γMAPK and calcium calmodulin protein kinases playing key roles. 30 Although high-intensity interval training raises both SIRT1 and PGC-1α levels, 31 low-volume sprint interval exercise in cyclists appears to achieve similar benefits. 32 Intriguingly, recent work suggests that the UPR plays a key role in skeletal muscle adaptation to exercise. This effect depends on PGC-1α cooperating with ATF6α to induce the UPR. 33 Given the role of the UPR to mediate organismal stress signaling, 10 PGC-1 activity may play an even more fundamental role in aging than currently appreciated.
Conclusion
The pioneering work of Sahin et al. 7 connecting replicative senescence with mitochondrial dysfunction, suggests that the master regulators of mitochondrial biogenesis PGC-1α and PGC-1β play an important role in aging-related pathologies. Increased expression of these genes undoubtedly explains the benefits of simple exercise. Conversely, reduction in their activity is contributing to the global pandemic of metabolic syndrome associated with sedentary lifestyles and overabundant nutrition. Development of new drugs that specifically augment PGC-1 activity may prove beneficial to prevent and reverse some of the detrimental effects of mitochondrial dysfunction seen with aging.