
Abstract
Homocysteine (Hcy) could induce amyloid β (Aβ) accumulation, synaptic dysfunction, and memory impairment as seen in Alzheimer disease (AD), the most prevalent neurodegenerative disorder, which affects more than 25 million people worldwide. Here we investigated the protective effect of hydroxysafflor yellow A (HSYA) on Hcy-induced Aβ accumulation, synaptic dysfunction, and learning and memory deficits. Rats were randomly divided into four groups: Control group, which received normal saline (NS); Hcy group, which received a daily vena caudalis injection of Hcy (400 μg/kg per day); Hcy+HSYA group, which received the same amount of Hcy plus 6 mg/kg per day HSYA intraperitoneally; and HSYA group, which received 6 mg/kg per day HSYA intraperitoneally for 2 weeks. Results showed that simultaneous supplementation of HSYA significantly attenuated Aβ accumulation, improved synaptic function, and reversed Hcy-induced cognitive impairment. Our data suggest that HSYA might be a promising therapeutic candidate for attenuating Hcy-induced AD-like pathological and behavioral deficits.
Introduction
A
Hydroxysafflor yellow A (HSYA), a major active chemical compound extracted from the traditional Chinese drug Carthamus tinctorius L., was approved clinically in 2005 by the Chinese State Food and Drug Administration. HSYA has been used for cardiocerebrovascular diseases. 6 HSYA exhibits numerous pharmacological properties, including anti-platelet aggregation, scavenging oxygen free radicals, lowering blood pressure and heart rate, and anti-inflammation and anti-tumor effects. 7,8 Moreover, HSYA showed neuroprotective effects on Aβ-induced neurotoxicity both in vitro and in vivo. 9,10
Homocysteine (Hcy) is an amino acid in the blood derived from the metabolism of methionine; it plays essential roles in cellular methylation for proteins, lipids, and DNA. Elevation of plasma Hcy is an independent risk factor for AD. 11,12 Mice with hyper-homocysteinemia exhibited a variety of the AD-like neuropathological changes, such as disruption of blood–brain barrier integrity, Aβ accumulation, and cognitive dysfunction. 13 –15 Previous studies reported that vena caudalis injection of Hcy-induced Aβ accumulation and tau hyper-phosphorylation. 16 These data suggest that hyper-homocysteinemia induced by Hcy could serve as a promising animal model for AD studies.
In the present study, we found that HSYA could rescue the impairment of memory retention ability, attenuate Aβ accumulation, and restore the synaptic dysfunction induced by Hcy.
Materials and Methods
Antibodies and chemicals
The specific primary antibodies in this study are listed in Table 1.
mAb, Monoclonal; pAb, polyclonal;
Animals and treatments
Male Sprague–Dawley rats (3 months old, 280±20 grams) were purchased from the Experimental Animal Center of Lanzhou University (Lanzhou, China). All rats were kept in cages under standard laboratory conditions—a 12-hr light–dark cycle and temperature at 22±2°C. All animal procedures were performed according to the Guide for Laboratory Animals Care and Use of Lanzhou University. Rats in the four groups were injected with Hcy (400 μg/kg per day) or saline in the same volume by vena caudalis for 2 weeks with or without an addition of HSYA (6 mg/kg per day) intraperitoneally 1 hr prior to the Hcy treatment. Animals were sacrificed 12 hr after the final injection.
Morris water maze
The spatial learning and memory ability was assessed using the Morris water maze. 17 The water maze was performed in a black cylindrical tank. The tank was equally divided into four quadrants. A transparent platform was submerged 1.5 cm below the water surface in the target quadrant. During the 6 days of spatial training, the platform was fixed. The rats were trained to find the platform using cues available in the testing room. A trial began by placing the rat into the water facing the wall of the pool at one of the starting points in the four quadrants. The animals were placed into the water facing the wall and allowed to find the hidden platform within 60 sec and stay on it for 20 sec. If the rat failed, then the rat was guided to the platform and allowed to remain there for 20 sec. On day 7, the swimming pathway and escape latency were examined. On day 9, the platform was removed and the percentage of time spent in the target quadrant and crossing times were tested. The swimming path was analyzed by using a computer-connected video tracking system (Bogazici Biyomedikal, Istanbul, Turkey).
Enzyme-linked immunosorbent assay
For quantitative analysis of Aβ levels in hippocampus, we performed a sandwich enzyme-linked immunosorbent assay (ELISA). Briefly, rat hippocampus was extracted and homogenized, and 200 μg of sample protein was used for ELISA. Aβ40 and Aβ42 in samples were captured with G2-10 and G2-11 respectively, and then were probed specifically with Biotin-WO2 (Abeta GmBH, Heidelberg, Germany). Samples were further developed with horseradish peroxidase (HRP)-NeutrAvidin (Pierce, IL), and the HRP activity was determined by using TMB (3,3′,5,5′-Tetramethylbenzidine) as substrate (Kirkegaard, MD). A standard curve in a linear range of 0.0–50.0 pmol/gram of Aβ (R 2=0.9912) was used for quantitative analysis of Aβ.
Measurement of long-term potentiation
The rats were anesthetized with urethane (1.2 grams/kg) and fixed on the stereotaxic instrument (Narishige, Tokyo, Japan). The temperature of the animal body was maintained at 37°C in a bath circulator. A small hole was drilled at the appropriate coordinates for vertical penetration of the stimulating and recording electrodes immediately after the skull was exposed. The stimulating electrode was placed in a perforant path with coordinates of anteroposterior (AP) −6.9 to −7.1 mm, dorsoventral (DV) −3.4 to −3.6 mm, and mediolateral (ML)±4.4 to±4.6 mm according to the rat brain atlas, and the recording electrode was fixed in CA3 at coordinates of AP −3.4 to −3.6 mm, DV −3.2 to −3.4 mm, and ML±3.4 to±3.6 mm. For each measurement, a stable baseline (±10% change, at least 20 min) was required before treatment of high-frequency stimulation (HFS). Long-term potentiation (LTP) was elicited by HFS consisting of four trains of 50 pulses delivered at 200 Hz with a 2-sec inter-train interval. LTP was assessed by the measurement of excitatory postsynaptic potential and population spike. 18
Western blot
Rat hippocampus were dissected and homogenized on ice in extraction buffer containing 50 mM Tris–HCl (pH 7.5), 1% (vol/vol) Triton X-100, 1% (wt/vol) deoxycholate, 150 mM NaCl, 0.1% (wt/vol) sodium dodecyl sulfate (SDS), 1 mM sodium orthovanadate (Na3VO4), 10 mM sodium fluoride (NaF), and 2 μg/mL each of leupeptin, aprotinin, and pepstatin A. The hippocampal extracts were separated by 10% SDS-polyacrylamide gel electrophoresis (PAGE) gels and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking in 3% non-fat milk for 30 min at room temperature, the membranes were incubated with specific primary antibodies at 4°C overnight and further incubated with horseradish peroxidase (HRP)-linked secondary antibodies. The blots were visualized by electrochemiluminescence (ECL; Pierce, IL). The optical density was analyzed by Kodak Digital software (Eastman Kodak, CT).
Analysis of plasma Hcy
Rats were anesthetized with 6% chloral hydrate (400 mg/kg) intraperitoneally. Plasma Hcy level was detected by using fluorescence high-performance liquid chromatograph (HPLC), as described previously. 19 Briefly, 60 μL plasma sample was mixed with 30 μL of 10% trichloroethyl phosphate (Sigma, NY) in 0.25 M borate buffer (pH 10.5). A 200-μL amount of 0.5 M perchloric acid containing 0.5 mM EDTA-Na2 was added. The homogenized samples were centrifuged at 13,000×g for 15 min. To 30 μL of the supernatant, a mixture consisting of 30 μ of 7-fluorbenzo-2-oxa-1,3-diazle-4-sulfonic acid (4.25 mM in 0.1 M borate buffer [pH 10.5] containing 2 mM of EDTA) and 20 μL of 0.5 M NaOH were added. A 10μL sample of the supernatant was analyzed by fluorescence HPLC at an excitation of 385 nm and emission 515 nm. Samples were separated using a reversed-phase column (Thermo, FL). Analysis was performed under isocratic conditions at a flow rate of 1.2 mL/min for 15 min. A standard curve in a linear range of 0.0–40.0 μM of Hcy (R 2=0.9942) was used for quantitative analysis of Hcy.
Statistical analysis
All results were expressed as means±standard deviation (SD) and analyzed using SPSS 16.0 statistical software (SPSS, IL) by one-way analysis of variance (ANOVA) procedure followed by a least significant difference (LSD) post hoc test. p<0.05 was set as statistically significant.
Results
HSYA arrested Hcy induced learning and memory deficits
The Morris water maze test was used to investigate the protective effect of HSYA. The rats were injected with 400 μg/kg per day of Hcy or normal saline (NS) for 2 weeks with/without an addition of HSYA (6 mg/kg per day) intraperitoneally 1 hr prior to Hcy treatment. Rats were trained in the Morris water maze followed Hcy treatment. The latency to search for the hidden platform was much longer in rats injected with Hcy compared with the NS group for both the training and memory tests (Fig. 1A, B). On day 9, decreased crossing times and less time spent in the target quadrant were found in the Hcy-treated rats (Fig. 1C, D). A supplement of HSYA efficiently rescued the Hcy-induced learning and memory deficits.
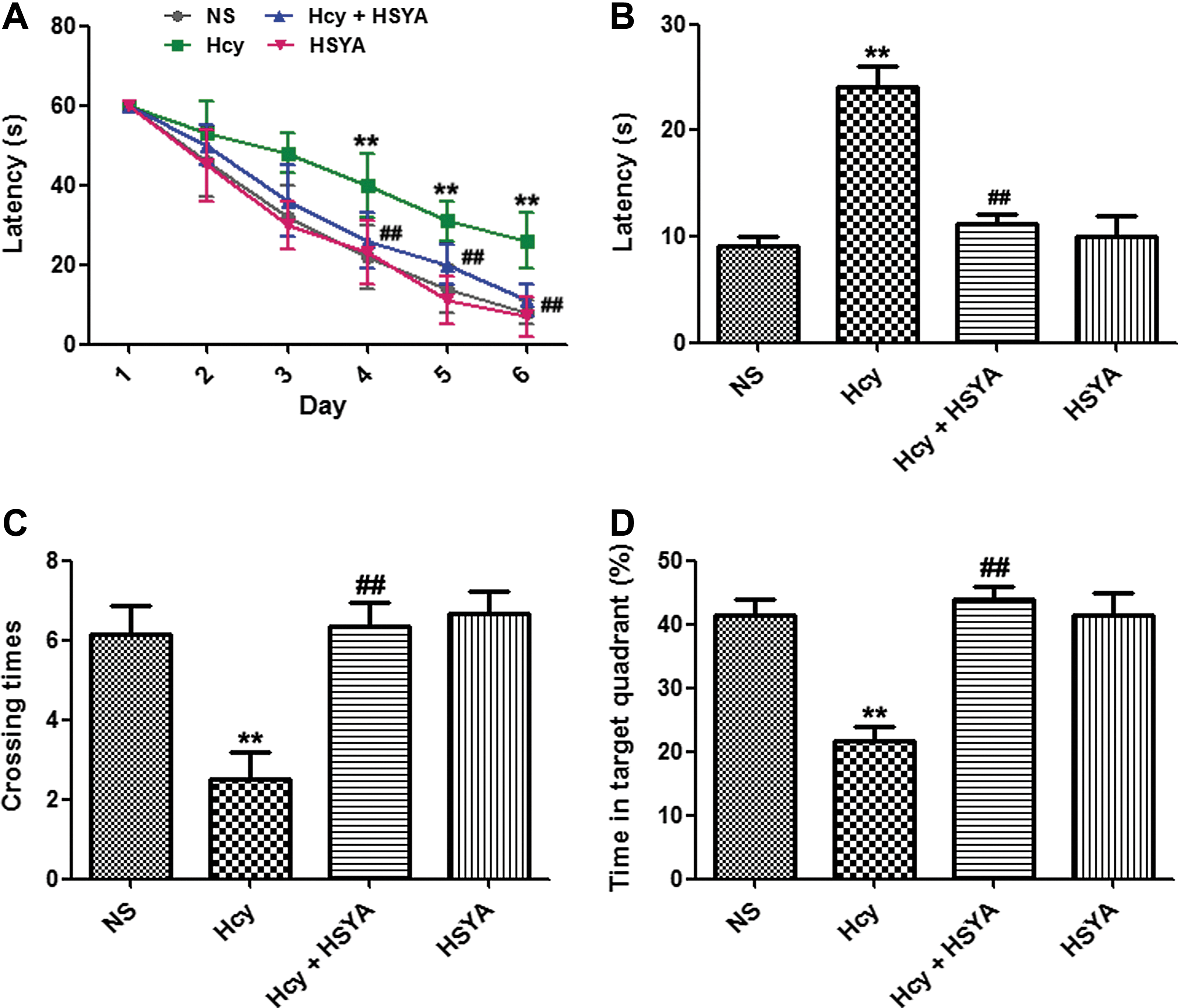
Hydroxysafflor yellow A (HSYA) arrested homocysteine (Hcy)-induced learning and memory deficits. (
HSYA attenuated Hcy induced Aβ accumulation
As reported previously, vena caudalis injection of Hcy for 2 weeks could induce Aβ accumulation. 16 We explored the effect of HSYA on Aβ accumulation in the brain hippocampus. Hcy increased both Aβ40 and Aβ42 levels in hippocampus, whereas HSYA treatment reversed the effect of Hcy (Fig. 2A, B). To further investigate the possible underlying mechanisms, we detected the expression of α-secretase, ADAM10, β-secretase, BACE1, and PS1 by western blotting. Supplementation of HSYA attenuated the elevation of PS1 induced by Hcy, whereas expression of ADAM10 and BACE1 were not obviously changed (Fig. 2C). To confirm the role of Hcy, we detected the plasma level of Hcy by fluorescence HPLC. A remarkable elevation of Hcy was observed after the injection (Fig. 2D).
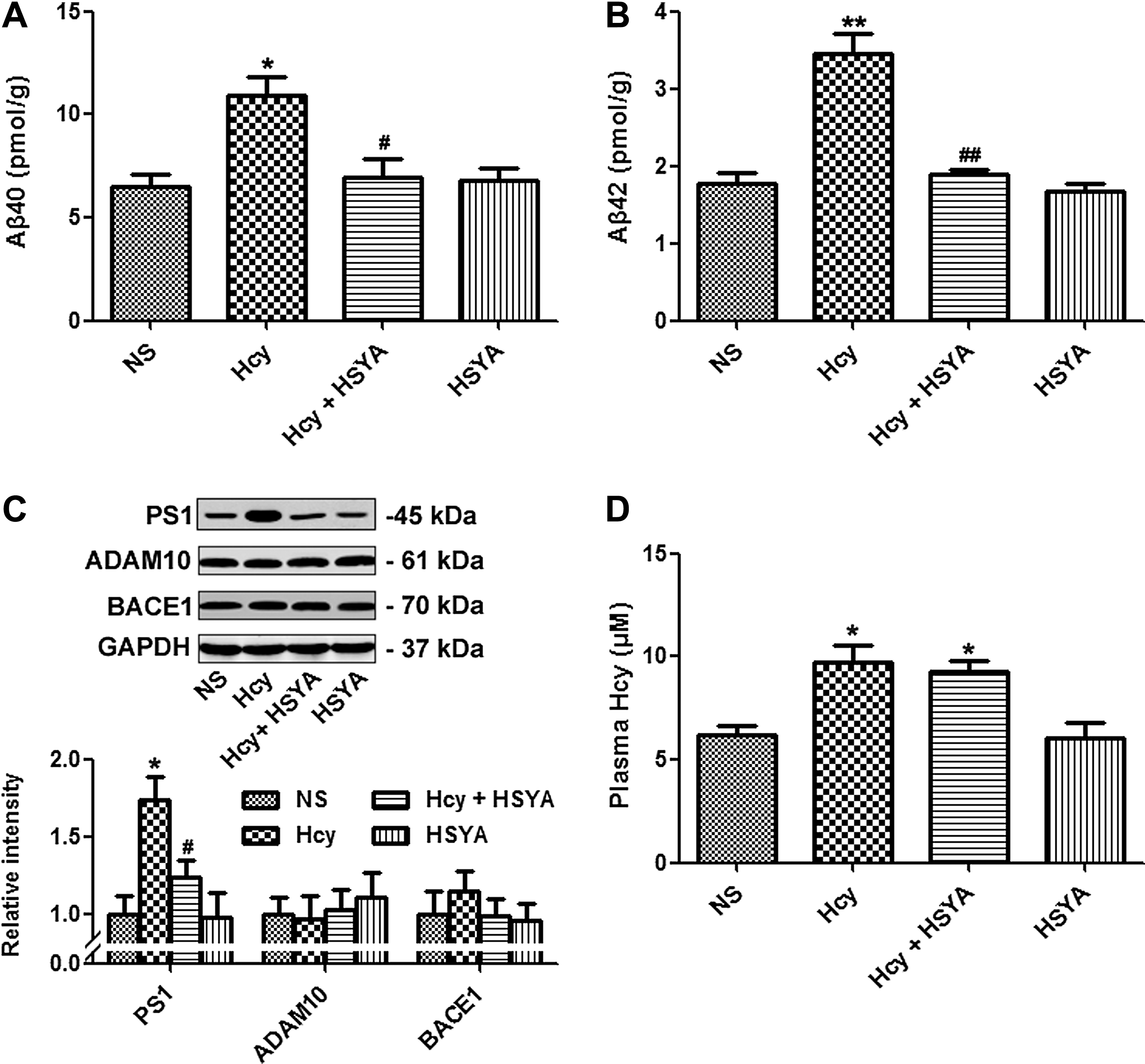
Hydroxysafflor yellow A (HSYA)-attenuated homocysteine (Hcy)-induced Aβ accumulation. Hcy (400 μg/kg per day) was injected with or without addition of HSYA (6 mg/kg per day) for 2 weeks. Rats were sacrificed, and Aβ40 and Aβ42 levels in hippocampus were measured by enzyme-linked immunosorbent assay (ELISA) (
HSYA rescued Hcy induced apoptosis
To examine whether HSYA could prevent the apoptosis induced by Hcy, we measured the expression of important molecules involved in the apoptosis pathway. Hcy increased expression of the pro-apoptosis molecule Bax, while inhibiting expression of the anti-apoptosis molecule Bcl-2. HSYA supplementation significantly reversed apoptosis induced by Hcy. The inhibitory effect of HSYA on apoptosis was supported by decreased level of the poly(ADP ribose) polymerase (PARP) cleavage product as well (Fig. 3A, B). These data suggest that HSYA could rescue Hcy-induced apoptosis.
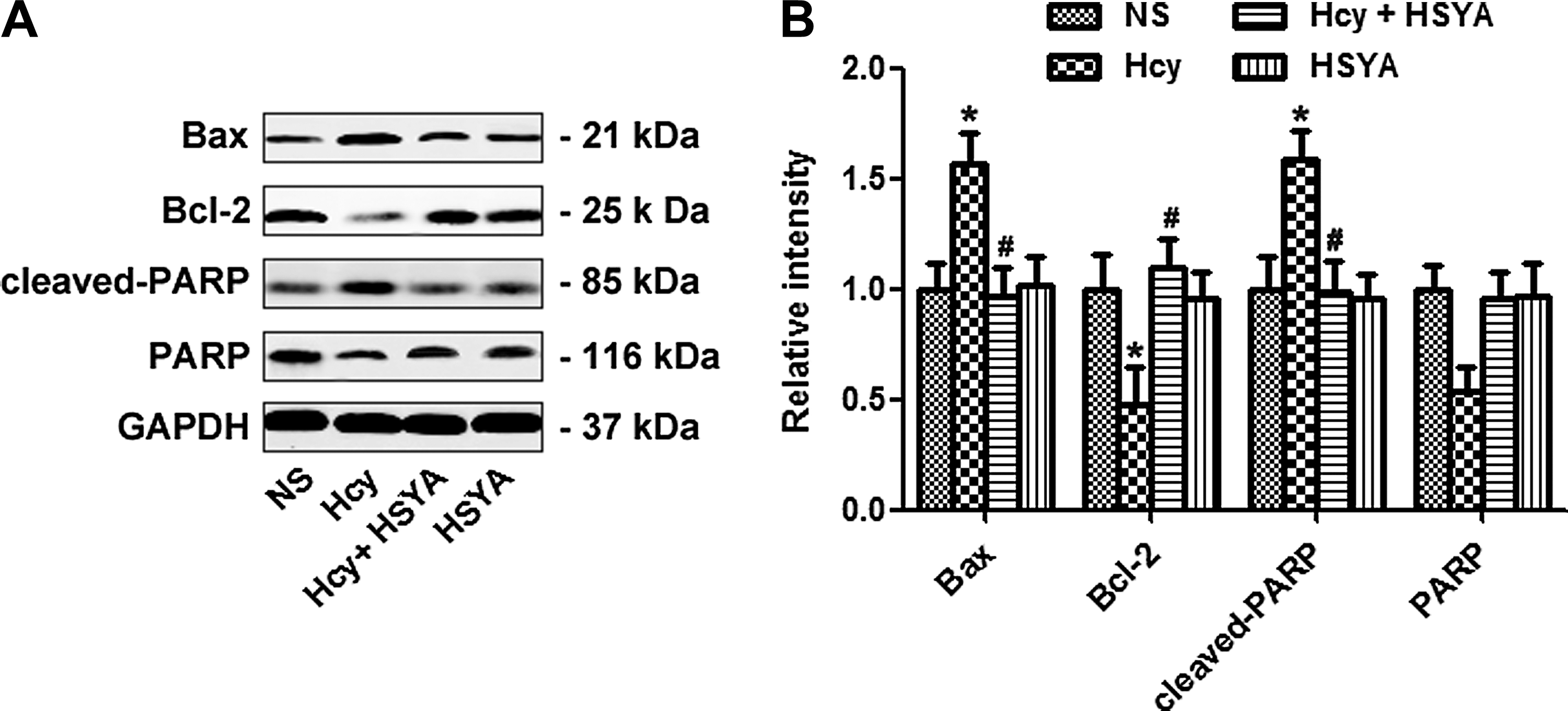
Hydroxysafflor yellow A (HSYA) rescued homocysteine (Hcy)-induced apoptosis. Apoptosis was assessed by measuring the expression of Bax, Bcl-2, and cleaved poly(ADP ribose) polymerase (PARP) (
HSYA reversed Hcy induced synaptic deficits
Synaptic plasticity is essential for learning and memory, and we determined the synaptic transmission by recording LTP. We found that in Hcy-treated rats, the slope and amplitude of field excitatory postsynaptic potential (fEPSP) increased to ∼1.3-fold and 1.4-fold, much lower than the NS group. Adding HSYA restored the amplitude and slope of fEPSP (Fig. 4A–C). These data show that HSYA could efficiently rescue the synaptic deficits induced by Hcy.

Hydroxysafflor yellow A (HSYA) reversed homocysteine (Hcy)-induced synaptic deficits. Hcy (400 μg/kg per day) was injected with or without addition of HSYA (6 mg/kg per day) for 2 weeks. Long-term potentiation (LTP) was detected in rats. (
Discussion
AD, which is characterized pathologically by numerous intracellular neurofibrillary tangles and intercellular senile plaques in the affected brain regions, is a specific and the most prevalent form of dementia in the elderly. Until now, the upstream factors triggering the formation of plaques were poorly understood. More than 40% of AD patients showed high Hcy levels in the plasma, displaying more rapid neural atrophy than those with normal Hcy levels. 20 Recently Ray et al reported that both AD and vascular dementia (VAD) subjects showed markedly elevated serum Hcy compared to control subjects. 21 Moreover, Chacón et al. found that elevated total Hcy (tHcy) was significantly associated with dementia, and increased tHcy was a significant risk factor in older (>66 years), but not in younger (55–66 years), subjects in a Venezuelan population that had high prevalence of both increased tHcy and dementia. 22 In AD, Aβ accumulation plays an essential role and co-exists with varying degrees of cerebrovascular dysfunctions. The elevated serum Hcy in AD subjects was thought to contribute to the genesis of the vascular pathology of AD. 23 Numerous studies had been performed to explore the direct neurotoxic effect of Hcy using cultured SHSY5Y cells, primary hippocampal neurons, rat brain cortical slices, and rat in vivo. 24 –26 Hyper-homocysteinemia was correlated with brain amyloidosis, Tau phosphorylation, electrophysiological properties, and cognitive impairment. 16,25,27 As a result, high Hcy has been proposed to be an independent risk factor of AD. Attenuating the elevated plasma Hcy level might be a potential strategy for AD therapy. 28 In the present study, we found that supplementation of HSYA could effectively rescue Hcy-induced learning and memory deficits, Aβ accumulation, and synaptic dysfunction in rats. Our data suggest that HSYA might serve as a potential candidate for AD therapy.
It is well known that Aβ is produced when APP is sequentially cleaved by β- and γ-secretases. 29 PS1 is the catalytic subunit of the β-secretase, and exogenous over-expression of PS1 stimulated Aβ production. 30,31 Previous studies demonstrated that Hcy could increase PS1 expression and promote Aβ production. 32 In this study, we found that HSYA significantly decreased both Aβ40 and Aβ42 levels partially through suppressing PS1 expression, which was elevated by Hcy. Unlike previously reported agents, such as VitB12/folate and betaine, HSYA did not affect the plasma Hcy level, suggesting that HSYA might work to counteract the downstream molecules affected by Hcy, or it might function competitively with Hcy and restore the effect of Hcy. Elevated plasma Hcy could induce Aβ accumulation, and HSYA might play a protective role in Hcy-induced Aβ accumulation partially by suppressing PS1 expression.
Emerging evidence showed Aβ deposition could increase pro-apoptosis factor expression, while suppressing anti-apoptosis molecule expression. In an Hcy-induced AD-like rat model, increased apoptosis was observed, as shown by increased expression of pro-apoptosis Bax expression and PARP cleavage. Hcy also decreased expression of the anti-apoptosis molecule Bcl-2. Supplementation of HSYA could efficiently rescue Hcy-induced apoptosis. Our in vivo study was consistent with previous in vitro studies. 9,33
Synaptic plasticity, which was disrupted in AD patients and AD animal models, could be assessed by LTP and was a prerequisite of learning and memory. 34,35 By detecting the alterations of EPSP slope and PS amplitude, well-accepted parameters for LTP measurement in vivo, we found that Hcy injection inhibited LTP in rat hippocampus, whereas supplementing with HSYA ameliorated the inhibitory effect of Hcy on LTP.
Taken together, we demonstrated in the present study that supplementation of HSYA significantly rescued Hcy-induced behavior impairment, Aβ accumulation, apoptosis, and synaptic dysfunction in rats. Our data suggest that HSYA might be a promising therapeutic candidate for attenuating Hcy-induced AD-like pathological and behavioral deficits.
Footnotes
Author Disclosure Statement
The authors have no conflict of interest to declare.