
Abstract
A fast and efficient method for purification of dihydromyricetin (3,5,7,3′,4′,5′-six hydroxy-2,3-dihydro flavonol; DMY) from Ampelopsis grossedentata was created by crystallization eight times at 25°C, and a purity of 98% was finally achieved. The purified DMY exhibited high oxygen radical absorbance capacity (ORAC) (30.21 μmol Trolox equiv/mg) and strong 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical scavenging activity (half-maximal inhibitory concentration [IC50]=0.235 μg/mL). The addition of DMY could also effectively attenuate 2,2′-azobis (2-amidinopropane) dihydrochloride (AAPH)-induced human erythrocyte hemolysis and cupric chloride (CuCl2)-induced human plasma lipid peroxidation via inhibition of intracellular reactive oxygen species (ROS) generation. It was also found that DMY (>12 μg/mL) treatment significantly inhibited intracellular malondialdehyde (MDA) formation. Meanwhile, DMY treatment significantly inhibited the obvious increase of anti-oxidant enzymes levels (superoxide dismutase [SOD]; glutathione peroxidase [GPX], and catalase [CAT]) induced by AAPH radicals, suggesting that stress defense mechanisms are associated with protection of DMY against intracellular oxidation.
Introduction
A
Over the last few years, considerable attention has been focused on the anti-oxidant activity of DMY. 11,12 So far, these studies have been based mainly on chemical analytical methods. In this study, DMY was first purified by recrystallization eight times. The protective effects of DMY against oxidation were evaluated by 2,2′-azobis(2-amidinopropane) dihydrochloride (AAPH)-induced erythrocyte hemolysis and cupric chloride (CuCl2)-induced oxidation of plasma assays as well as by chemical assays. This work is the first attempt to investigate the intracellular anti-oxidant properties of DMY with the hope of obtaining a better understanding of the mechanisms involved with DMY to quench intracellular reactive oxygen species (ROS).
Materials and Methods
Materials and chemicals
A. grossedentata was kindly provided by the Food Biochemistry Laboratory of South China University of Technology (Guangzhou, China). AAPH and 2,2-diphenyl-1-picrylhydrazyl (DPPH) were purchased from Sigma-Aldrich (St. Louis, MO). A malondialdehyde (MDA) kit was obtained from Nanjing Jiancheng Institute of Biotechnology (Nanjing, China). Assay kits for determination of cellular glutathione peroxidase (GPX), superoxide dismutase (SOD), ROS, bicinchoninic acid (BCA), and catalase (CAT) were purchased from the Beyotime Institute of Biotechnology (Shanghai, China). The ultra-pure water used in all experiments was prepared by a Milli-Q water purification system (Millipore, Schwalbach, Germany).
Purification of DMY by recrystallizations
The leaves and tender stems powder of dried A. grossedentata were continuously extracted with distilled water at 100°C for 1 hr. The supernatants were filtered at 80–90°C and crystallized at room temperature for 12 hr. Recrystallizations were carried out eight times in water to purify DMY on the basis of its different solubility in cold or boiled water. The purity of DMY was identified by high-performance liquid chromatography (HPLC; Symmetry C18 column 3.5×150 mm, Waters Co., USA) using a mobile phase consisting of acetonitrile–water–acetic acid (1:9:0.1, vol/vol; pH 3.5). The elution was conducted at a flow rate of 1.0 mL/min at a column temperature of 25°C with an injection volume of 10 μL. Finally, the effluent was monitored at 293 nm.
Scanning electron microscopy imaging
The morphology of DMY samples was examined using a scanning electron microscope (SEM; Hitachi TM3000, Hitachi Ltd., Tokyo, Japan). The samples of DMY were placed on a specimen holder with the help of double-sided Scotch Tape and sputter-coated with gold (5 min, 2 mbar). The treated blood samples were spread onto freshly cleaved mica to create a blood film with a single layer of red blood cells and fixed with 2.5% glutaraldehyde. Then the blood film was pasted onto a specimen holder. Finally, both of the treated samples of DMY and blood film were transferred to the SEM and examined.
Chemical anti-oxidant capacity assays
Oxygen radical absorbance capacity (ORAC) assay 13
Briefly, a volume of 20 μL of blank Trolox standard or samples (dissolved in 75 mM phosphate-buffered saline [PBS], pH 7.4) was added to a 96-well black plate. Then 20 μL of 70 nM fluorescein was added to each well and incubated at 37°C for 20 min. After incubation, 140 μL of 12.8 mM AAPH was added immediately to each well, and the plate was placed in a Fluoroskan Ascent™ Microplate Fluorometer (Thermo Electron Corporation, Vantaa, Finland) with 485-Pexcitation and 528-Pemission filters for fluorescence measurement. The plate was shaken automatically to mix the reagents before the first reading, and the fluorescence was monitored every 3 min for a total of 108 min. All of the samples were prepared in triplicate. A standard curve was established by testing different concentrations of Trolox. According to the normalized curves, the area under the curve (AUC) was calculated as: AUC=(0.5+f 1/f 0+f 2/f 0+f 3/f 0+… +f 60/f 0)×CT, 14 where f 0 stands for the initial fluorescence reading at 0 min and CT stands for the cycle time in minutes. The net AUC corresponding to each sample was calculated by subtracting the AUC of the blank. The regression equation of net AUC and Trolox concentration was established, and the ORAC values were represented as μM TE (Trolox equivalent)/mg of anti-oxidant.
DPPH radical scavenging activity assay
The DPPH scavenging activity of DMY was measured by colorimetric method. 15 Briefly, an aliquot of 0.1 mL of diluted DMY was mixed with 2.9 mL of 6×10−5 M DPPH solution (dissolved in 50% ethanol). The mixture was mixed thoroughly and incubated in the dark for 60 min at room temperature. The variation of the absorbance was measured at 517 nm using a spectrophotometer (UV2550, SHIMADZU, Kyoto, Japan). All tests were done in triplicate.
The radical scavenging capacity was calculated as follows:
Anti-oxidant assays based on cell models
Peroxyl radical–mediated hemolysis assay
The effect of DMY on the inhibition of erythrocyte hemolysis was measured. 16 Blood was obtained from healthy adult male volunteers. Erythrocytes and plasma were separated by centrifugation at 1200×g for 10 min at 4°C. Then the erythrocytes were washed with PBS (pH 7.4) for three times and re-suspended in PBS buffer to a hematocrit level of 20%. An aliquot of 0.2 mL of erythrocyte suspension was mixed with different concentrations of DMY (0.2 mL). PBS buffer was used as control. The mixture was shaken gently and incubated at 37°C for 20 min before the addition of 0.4 mL of 200 mM AAPH. The mixture obtained was incubated for another 2 hr at 37°C and then diluted with 8 mL of PBS buffer. As a comparison, the complete hemolysis group was diluted with 8 mL of ultrapure water. Samples were centrifuged at 1200×g for 10 min at 4°C. The absorbance of the supernatant was determined at 540 nm using a spectrophotometer (UV2550, SHIMADZU, Kyoto, Japan). The percentage of the hemolysis inhibition was calculated as follows: % inhibition=(1 − Ac/As)×100%, where Ac stands for the absorbance of control in PBS buffer and Ac stands for the absorbance of DMY with difference concentrations.
The morphology of the erythrocytes with or without the treatment of DMY samples was examined by a SEM (Hitachi TM3000, Hitachi Ltd., Tokyo, Japan), as described above. The treated blood samples were spread onto freshly cleaved mica to create a blood film with a single layer of red blood cells, fixed with 2.5% glutaraldehyde, transferred to the SEM, and examined.
Plasma oxidation assay
Plasma samples were separated from whole blood from healthy adult male volunteers by centrifugation at 1200×g for 10 min at 4°C and diluted 40-fold with PBS, aliquoted, and stored at 4°C before use. Plasma (0.2 mL) was mixed with 0.2 mL of DMY of different concentrations in centrifuge tubes and shaken gently while being incubated at 37°C for 20 min. Then, 0.4 mL of 200 μM CuCl2 was added to each tube, mixed thoroughly, and incubated at 37°C for another 2 hr. The formation of diene was monitored at 245 nm every 10 min.
Intracellular ROS level assay
The intracellular ROS level was determined using a ROS Assay Kit (Beyotime Institute of Biotechnology, Haimen, Jiangsu, China). Briefly, the erythrocytes after DMY treatment were collected by centrifugation at 1200×g for 10 min at 4°C and washed three times with PBS. Then the cells were resuspended in 10 μM 2′,7′-dichlorofluorescein diacetate (DCFH-DA, a fluorescent probe) and incubated at 37°C for 20 min. The DCFH-DA was oxidized by ROS and de-esterified to dichlorodihydrofluorescein to produce the fluorescent compound dichlorofluorescein in vivo. The fluorescence intensity of cells was measured with the Fluoroskan Ascent™ Microplate Fluorometer (Thermo Electron Corporation, Vantaa, Finland) (ex/em, 488/525 nm).
Lipid peroxidation and GPX, SOD, and CAT levels assay
Cells were harvested after treatment by centrifugation and washed with PBS as described above. Then the cells were lysed in 600 μL of cold Milli-Q water and stored at −80°C before analysis. The protein concentration was determined by a BCA Protein Assay Kit (Beyotime Institute of Biotechnology, Shanghai, China). Lipid peroxidation was monitored by measuring the produced MDA level using a Microamount MDA kit based on the thiobarbituric acid (TBA) reactivity. GPX, SOD, and CAT activities were measured by a Cellular Glutathione Peroxidase Kit, total Superoxide Dismutase Assay Kit, and Catalase Assay Kit (Beyotime Institute of Biotechnology, Shanghai, China), respectively. All these measurements were carried out by strictly following the manufacturer's instructions.
Statistical analysis
All of the experiments were carried out in triplicate. The data were expressed as means±standard deviations (SDs) and analyzed by SPSS 17.0 software (SPSS, Inc., Chicago, IL), and the mean values were considered significantly different at p<0.05.
Results and Discussion
Purification of DMY by crystallization
Effect of crystallization temperature
The crystallization of DMY was carried out at different temperatures, 4°C and 25°C, respectively, and the SEM micrographs were shown in Fig. 1. It was found that when the crystallization was conducted at 4°C (Fig. 1a), a large number of irregular small crystals were formed, but the crystals had a wide variety of sizes. Most of the crystals were very small in size and scattered everywhere. As the reaction temperatures increased to 25°C (Fig. 1b), it was observed that more regular structures were formed and no obvious cuboid aggregates appeared. The surface of the DMY crystal obtained was seen to be smooth, and the structures were found to be composed of cuboid aggregates of a larger size of crystals. The result suggests that crystallization temperature had an obvious effect on the crystal structure of DMY and, obviously, DMY obtained under 25°C looks better than that treated at 4°C.

Scanning electron microscope (SEM) images of dihydromyricetin (DMY) crystallized at different conditions: (
Effect of crystallization times
The crystallization of DMY was repeated for two, four, six, and eight times, respectively. When crystallized twice (Fig. 1c), the DMY obtained formed bits and pieces of solid rectangular crystals and assembled in cluster form. As the crystallization times increased, the DMY crystals obtained became more regular and the formed cuboid aggregates tend to be scattered (Fig. 1d and 1e). After crystallization eight times, the structure of the DMY crystal obtained was more regular (Fig. 1f). This indicated that the treatment times play an important role in modulating the morphology of the crystals. To test the purity of the DMY obtained after crystallization eight times, the samples were freeze-dried, re-dissolved in methanol, and analyzed by HPLC on a Symmetry C18 column (3.5×150 mm, Waters Co., USA). As shown in Fig. 2, a good separation of DMY with the purity of 98% was achieved. As compared with the previous reports about DMY purification using alcohol as a solvent, we found that using water as a solvent could effectively enhance the purity of DMY.
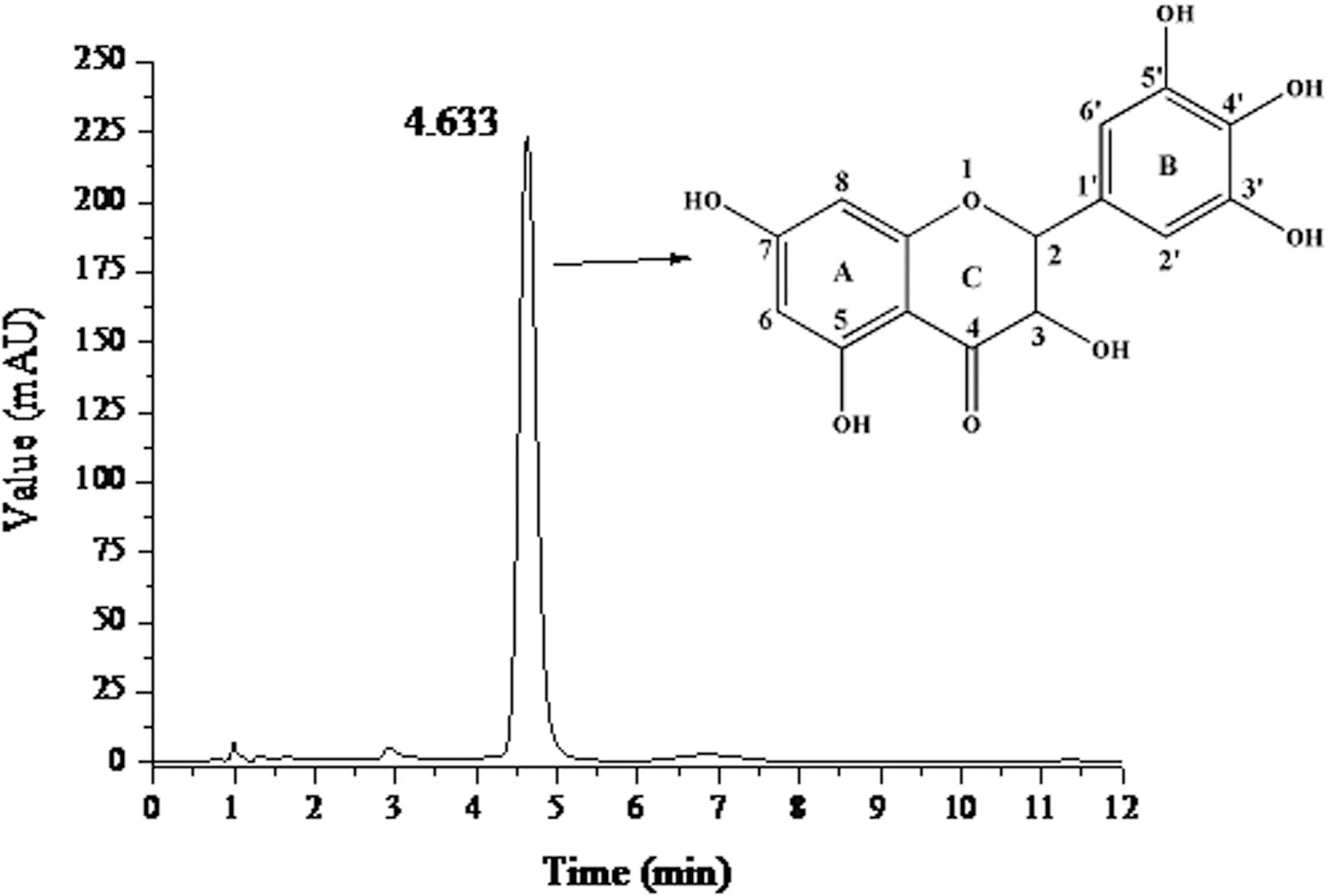
High-performance liquid chromatography (HPLC) analysis of purified dihydromyricetin (DMY) on a Symmetry C18 column (3.5×150 mm) and its chemical structure.
Anti-oxidant activities of DMY by chemical assays
Generally, there are two categories of chemical anti-oxidant capacity assays—hydrogen atom transfer (HAT) reaction–based assay and single electron transfer (SET) reaction–based assay. 11 In the present study, an ORAC assay based on HAT reaction and a DPPH radical scavenging assay based on SET reaction were conducted to evaluate the anti-oxidant capacity of DMY.
The ORAC assay has been widely used to detect anti-oxidant activity in food and biological systems. It measures the inhibition of peroxyl radical (LOO•)-induced oxidations, as free radicals can decrease the fluorescence emission of the fluorescence probe. In the present study, the ORAC value of DMY was calculated to be 30.21 μmol Trolox equiv/mg, indicating a strong anti-oxidant capacity of DMY. This might be due to the special structure of DMY, which has six hydroxyl groups attaching to the C3, C5, C7, C3′, C4′, and C5′ positions of DMY (Fig. 2). The most active H-donating hydroxyl group was the one attached to C4′, 5 which contributed much to its high ORAC value.
The DPPH radical scavenging activity is a typical assay method based on the SET reaction. DPPH is long-lived nitrogen radical, and the assay is based on the decrease in absorbance at 517 nm caused by the reduction of DPPH radicals in the presence of DMY. As shown in Fig. 3a, the half-maximal inhibitory concentration (IC50) of DMY-inhibited DPPH radical was 0.235 μg/mL, indicating an efficient free radical scavenging activity of DMY under the hydrophobic condition. According to the structure of DMY (Fig. 2), the hydroxyl group (on the C ring) of the 3-position (flavonols) should also have positive effects on its radical scavenging activity. 17
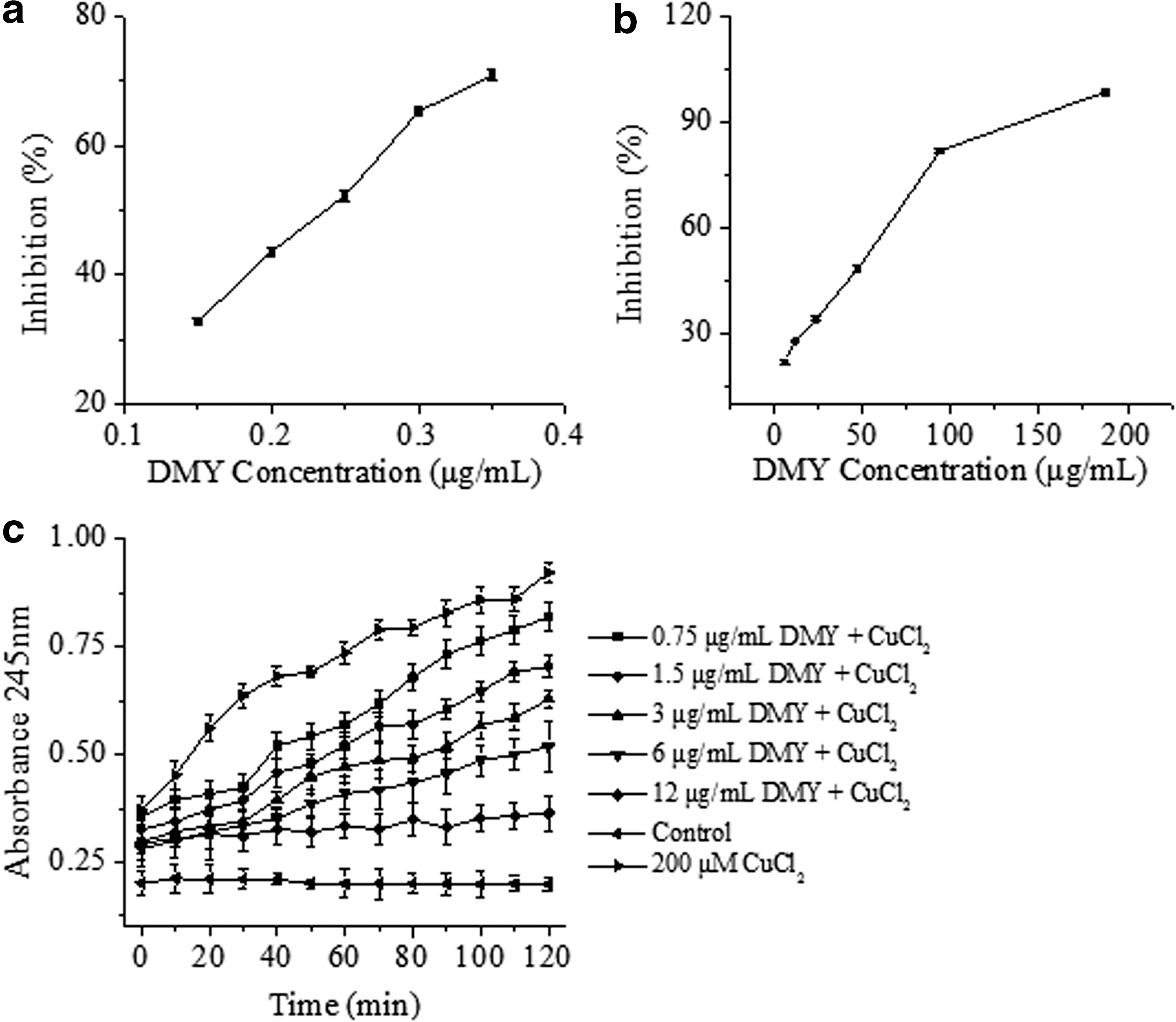
Anti-oxidant activities of dihydromyricetin
Anti-oxidant activities of DMY on the basis of cell models
Attenuation of erythrocyte hemolysis and plasma oxidation by DMY
In vitro–cultured cell model systems are also valuable methods for anti-oxidant evaluation of DMY. In this study, the protection effects of DMY against oxidation were evaluated by erythrocyte hemolysis assay and plasma oxidation assay. For erythrocyte hemolysis assay, AAPH was used as a radical initiator, which could generate free radicals by thermal decomposition at physiological temperature. The free radicals produced would attack red blood cells inducing the peroxidation of lipids and proteins, damage the membrane, and ultimately lead to hemolysis. 18 Therefore, the inhibition rate of hemolysis can be regarded as the indirect evidence of the anti-oxidant activity of DMY. As shown in Fig. 3b, DMY showed dose-dependent protection effects on red blood cells against the hemolysis induced by AAPH. The inhibition rate reached to 98.7% at 188 μg/mL of DMY and the IC50 was found to be 56.4 μg/mL. A well-known anti-oxidant, vitamin C, was used as the positive control in this experiment with the inhibition rate of 96.9% at 1 mg/mL (data not shown). The results demonstrated that the anti-oxidant activity of DMY was even superior to that of vitamin C.
In addition, the morphological changes of erythrocytes after APPH-induced damage with or without DMY protection were examined by SEM. As shown in Fig. 4a, a normal erythrocyte maintains a rounded and biconcave shape with a smooth surface. After treatment with 200 mM AAPH for 2 hr, the cell membranes were destroyed and the surface of erythrocyte became rougher and spinous (Fig. 4b). However, the incubation of erythrocytes with 188 μg/mL of DMY significantly alleviated AAPH-induced damage of erythrocytes, and the erythrocyte regained its normal morphology (Fig. 4c). This further confirmed that DMY could effectively attenuate AAPH-induced oxidative stress in erythrocytes. In the plasma oxidation assay, the time course of CuCl2-induced conjugated diene formation at 245 nm upon exposure of plasma to CuCl2 is shown in Fig. 3c. It was found that 200 μM copper (II) ion treatment induced rapid lipid peroxidation and diene accumulation in the plasma, 19 whereas the addition of DMY with different concentrations (0.75, 1.5, 3.0, 6.0, and 12.0 μg/ mL) exhibited obvious inhibition of copper-dependent conjugated diene, with a concomitant protective effect against oxidative damage in plasma. A dose-dependent effect was also found because with the increase of DMY dosage, more effective inhibition capacity was observed.

Scanning electron microscope (SEM) micrographs of erythrocyte samples. (
DMY attenuates AAPH-induced ROS generation in erythrocytes
ROS includes a number of highly reactive molecules such as hydroxyl radical (OH•), superoxide anion (O2 • −), peroxyl radicals (ROO•), hydrogen peroxide (H2O2), singlet oxygen ( 1 O2), and peroxynitrite (ONOO− ), which are known as important mediators of intracellular signaling palthways. 20 However, the overproduction of ROS could lead to oxidative damage, cell dysfunction, and eventually cell apoptosis or necrosis. 21 ROS generation is regarded as a biomarker of intracellular oxidative stress, and the protection effect of anti-oxidants against cell oxidation can be evaluated by their ability to decrease the level of ROS generation in vivo. 22
In the present study, the protection effects of DMY on AAPH-induced ROS generation in erythrocytes were evaluated, and a fluorescein-labeled dye (DCFH-DA) was applied to detect the intracellular ROS level. As shown in Fig. 5, 200 mM of AAPH treatment induced a significant increase of ROS levels in erythrocytes, which is consistent with the previous study. 23 The addition of DMY (6, 12, 24, 48, 96, and 188 μg/mL) could notably reduce AAPH-induced ROS generation. Furthermore, it was found that a single DMY (188 μg/mL) treatment was safe for erythrocytes without enhancing its intracellular ROS level, as compared with the control group. With these findings, it is reasonable to speculate that DMY could attenuate AAPH-induced human erythrocytes via inhibition of intracellular ROS generation.
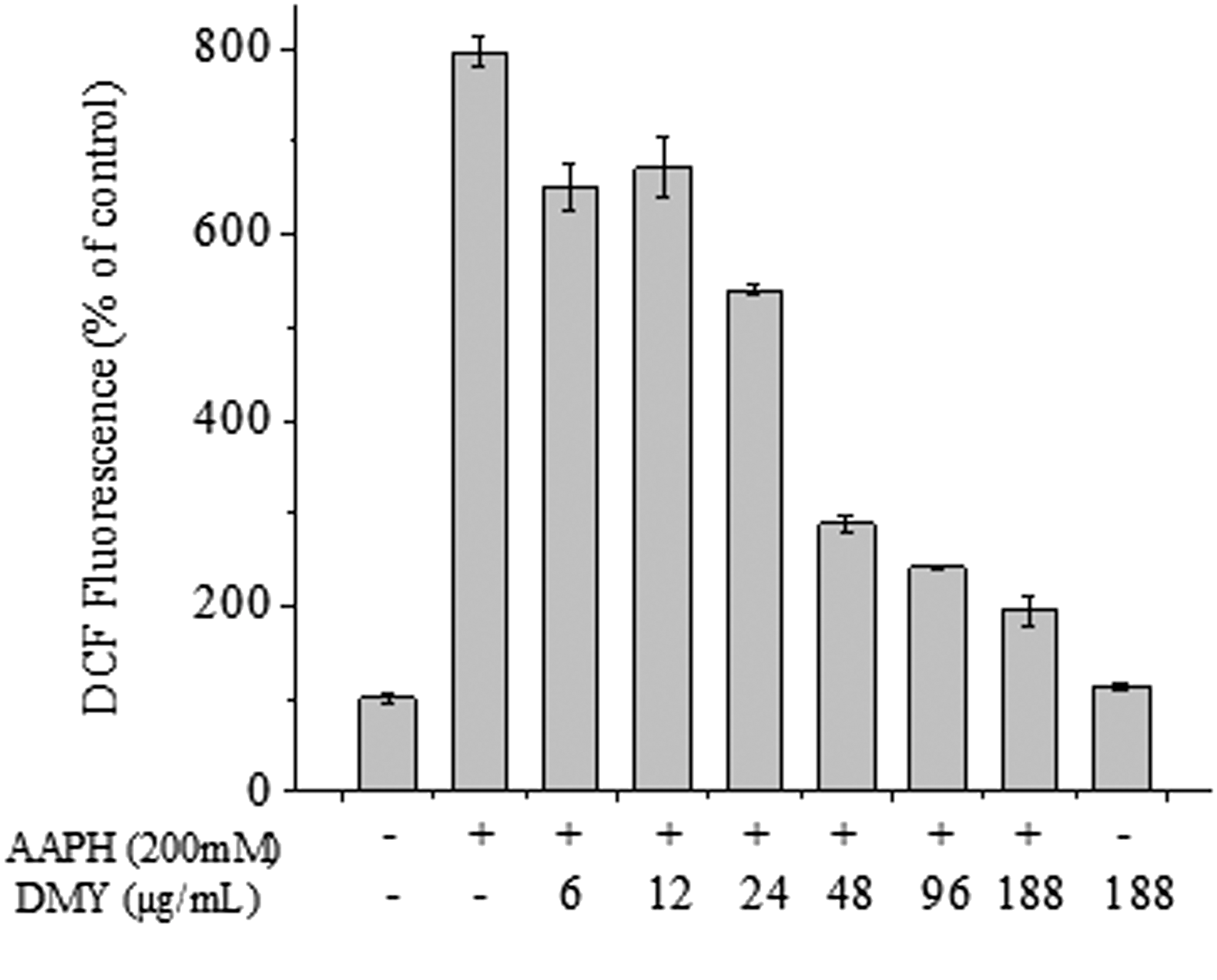
Effects of dihydromyricetin
DMY inhibits AAPH-induced MDA production and modulates intracellular anti-oxidant enzymes (GPX, SOD, and CAT)
Excess ROS generation leads to oxidation of lipids, carbohydrates, and proteins. 24 Lipid peroxidation results in the release of MDA, which is involved in tumor promotion, disruption of cellular metabolism, and cell membrane dysfunction due to its high reactivity. 25 As shown in Fig. 6a, AAPH treatment significantly increased the level of MDA in erythrocytes. The MDA content increased from 0.56 nmol/mg protein to 13.19 nmol/mg protein after treatment with 200 mM AAPH for 2 hr, indicating the occurrence of lipid peroxidation caused by AAPH-induced oxidative stress. When a different concentration of DMY was added, MDA formation was significantly inhibited, especially for high concentration (>12 μg/mL) treatments. Obviously, DMY could efficiently alleviate the AAPH-induced lipid peroxidation in erythrocytes. In addition, a single DMY treatment without AAPH addition would not cause the increase of MDA level in erythrocytes in comparison with the control group, indicating that DMY itself has no potential to induce MDA formation.
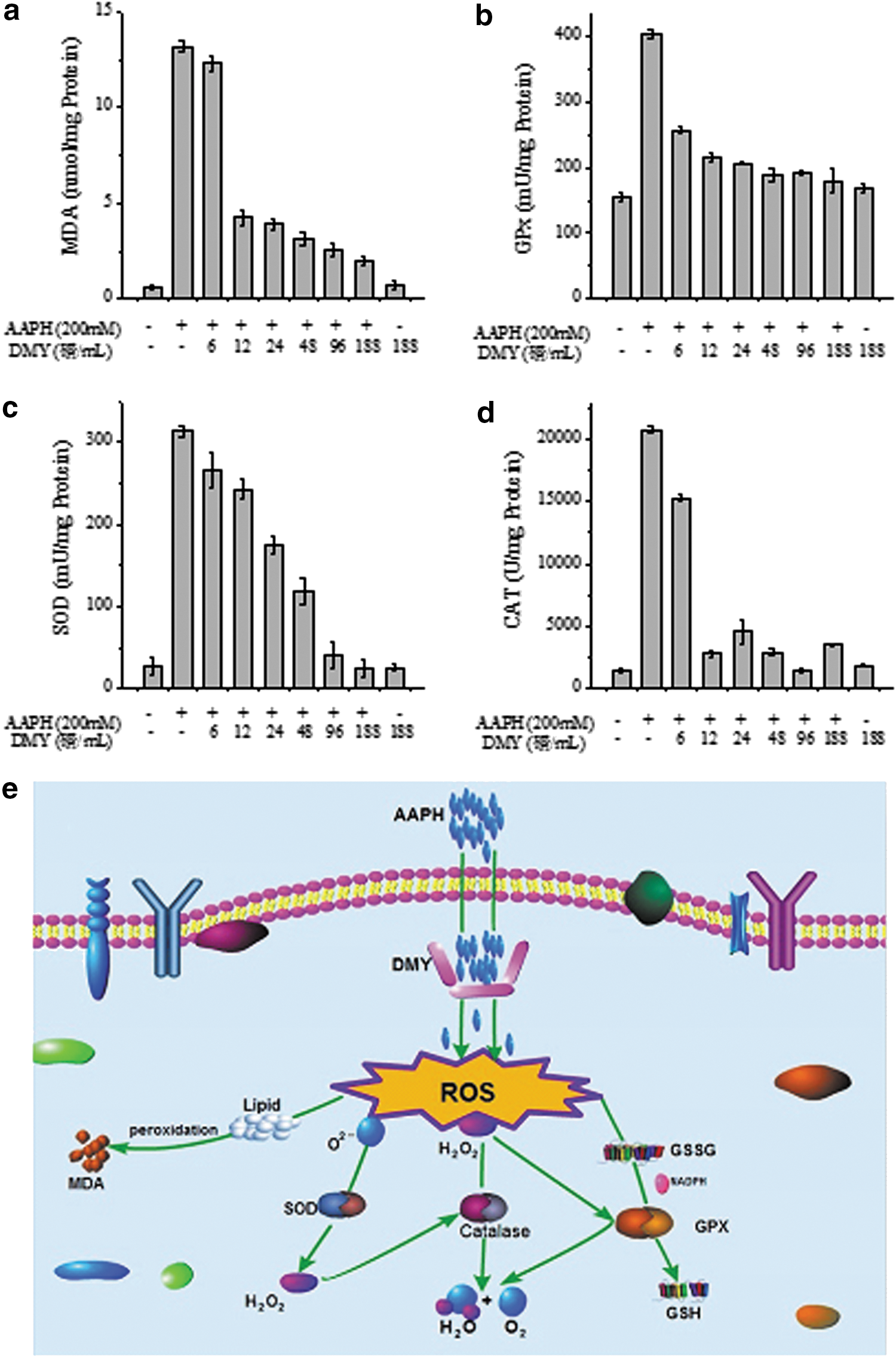
Changes of malondialdehyde (MDA) (
Overproduction of ROS resulted in oxidative stress; meanwhile, the intracellular anti-oxidant defense systems of cell itself could be activated to protect the cell against oxidative damage. 26 There are two major groups of cellular anti-oxidant systems, including non-enzymatic and enzymatic systems. 27 Non-enzymatic defense systems include some anti-oxidants such as glutathione (GSH), μ-carotenes, and vitamins A, C, and E. 28,29 In the present study, we focused on the intracellular defense systems consisting of SOD, CAT, and GPX, which can coordinate the elimination of free radicals by a series of chain reactions. 30 SOD is one of the most important anti-oxidant enzymes within the cell and quenches superoxide radical (O2 −) by converting it into O2 and H2O2. 31 H2O2 can be then reduced to H2O by CAT; otherwise H2O2 might react with transition metals and thereby be converted into •OH, the strongest radicals that can cause cell death. 28 The main role of GPX is to catalyze the oxidation of GSH; it can also reduce the content of H2O2 and ROO• in the process of GSH oxidation. 32
In present study, it was found that after 200 mM AAPH treatment for 2 hr, the SOD, GPX, and CAT activities were all significantly enhanced (Fig. 6b–d), indicating that the enzymatic anti-oxidant defense systems of the erythrocytes were activated by AAPH. The erythrocyte groups treated by DMY only were used as controls to evaluate the cytotoxicity of DMY on erythrocytes. As shown in Fig. 6a–d), the values for single DMY treatment without the AAPH addition group were quite similar to those of the negative control group (neither DMY nor AAPH treated), indicating that the working concentrations of DMY did not affect the viability of erythrocytes. The anti-oxidant system of the cells was built up through quenching of O2 − by SOD and further protection of CAT to convert the H2O2 obtained to H2O and thus prevent the formation of •OH (Fig. 6e). In this study, we found that the presence of DMY decreased GPX (Fig. 6b), SOD (Fig. 6c), and CAT (Fig. 6d) in the erythrocytes to a normal level. All of these results suggested that DMY had positive effects on erythrocytes against the oxidative damage.
In conclusion, we found that the effective purification of DMY from A. grossedentata could be achieved by crystallization eight times using water as the solvent, instead of using ethanol as the solvent as in traditional extraction process. The purified DMY exhibited a high ORAC value and strong DPPH radical scavenging activity, due to the contribution of a H-donating hydroxyl group attached to C4′ and the hydroxyl group (on the C ring) of the 3-position (flavonols) in the DMY structure. DMY could also effectively attenuate AAPH-induced oxidative stress in erythrocytes and plasma via inhibition of intracellular ROS generation. All of these findings suggest that DMY could be a potential candidate for the treatment of diseases in which ROS over-production acts as the etiological factor.
Footnotes
Acknowledgments
This study was supported by the National Key Technology R&D Program of China in the 12th 5-year period (no. 2012BAD33B11), the Fundamental Research Funds for the Central Universities (no. 2014ZZ0063 and 2013ZZ0061), and Guangdong Natural Science Funds for Distinguished Young Scholar (no. S2013050013954).
Author Disclosure Statement
All authors declare no conflict of interest.