
Abstract
Oxidative stress and decreased DNA damage repair in vertebrates increase with age also due to lowered cellular NAD+. NAD+ depletion may play a major role in the aging process at the cellular level by limiting (1) energy production, (2) DNA repair, and (3) genomic signaling. In this study, we hypothesize that it is not NAD+ as a cofactor in redox reactions and coenzyme in metabolic processes that has the ultimate role in aging, but rather the role of NAD+ in cellular signaling when used as substrate for sirtuins (SIRT1-7 in mammals) and PARPs [Poly(ADP-ribose) polymerases]. Both sirtuins and PARPs influence many transcription factors and can affect gene expression. As a signaling molecule, NAD+ is consumed in the reaction donating ADP-ribose and releasing nicotinamide (NAM) as a by-product. It seems that aging at the cellular level is associated with a decline of NAD+ and that NAD+ restoration can reverse phenotypes of aging by inducing cellular repair and stress resistance. Adequate intracellular NAD+ concentrations may be an important longevity assurance factor, while lowered cellular NAD+ concentration may negatively influence the life span.
Introduction
T
NAD+
NAD+ is a coenzyme involved in cellular redox reactions and is a substrate for NAD-dependent enzymes. The involvement of NAD+, nicotinamide phosphoribosyltransferase (NAMPT), and SIRT1 in the regulation of metabolism and aging was systematically presented by the group of Dr. Imai and named NAD World. 19 –21 The NAD+/NADH ratio might influence the formation of ROS and oxidative stress due to the regulation of the intracellular ATP production, redox state, and metabolic enzymes. 22,23 Changes in the NAD+ level and/or the NAD+/NADH ratio can influence the life span in yeast 24 by inducing DNA repair and increasing cell defense and by regulating diverse signaling pathways 23,25,26 and transcriptional events (for example, in pancreatic cells 24 ). With advancing age, NAD+ and nicotinamide mononucleotide (NMN) levels decline and NADH level increases. 2,27 The NAD+/NADH ratio plays an important role in regulating the intracellular redox state, and several enzymes involved in regulation of metabolism are regulated by the NAD+/NADH ratio. The NAD+/NADH ratio is an index of cellular reduction potential and is regulated by small changes in NAD+ concentration. 22,28 Aging seems to be promoted when the NAD+/NADH ratio is moved toward NADH. Namely, NADH is a competitive inhibitor of Sir2 29,30 and excessive amounts of NADH were reported to produce the state of reductive stress and to facilitate ROS formation by NADH-induced iron release from ferritin or from the electron transport chain. 31 –33 On the contrary, when the ratio is moved toward NAD+, prosurvival pathways are activated. 29 Namely, the ratio of NAD+/NADH regulates enhanced oxidative metabolism, DNA repair, stress resistance, and cell death. 22,24,34 NAD+ is involved in the aging process also through regulation of tankyrase activity (mediators of telomerase activity). 35
Increasing NAD+: Exercise, Caloric Restriction, and Ingestion of NAD+ Precursors and Intermediates
Increasing NAD+ biosynthesis with NAD+ intermediates or by other approaches could be a strategy for preventing and treating aging and age-associated diseases. 19,23,36,37 Aerobic exercise, caloric restriction (CR), fasting, and low-glucose availability increase the NAD+ levels (probably through an increase in the NAMPT gene expression), increase mitochondrial and sirtuin activities, and lower the NADH levels. 38 –42 NAMPT is an enzyme essential for NAD+ biosynthesis, responsible for converting nicotinamide (NAM) to NMN, which is then converted to NAD+. 19,43 NMN is a key NAD+ intermediate. 44 Costford et al. 45 demonstrated that exercise increases skeletal muscle NAMPT expression and the amount of NAD+ and that NAMPT correlates with the amount of mitochondria. Exercise additionally increases the NAD+ level by reoxidizing NADH by mitochondrial respiration. 46,47 The beneficial effects of CR appear to be also NAD+ dependent as well as mediated by the NAD+-dependent silent information regulator (SIRT1/Sir2) activity. 34,48 Calorie restriction extends yeast's life span by lowering the level of NADH, since NADH is a competitive inhibitor of Sir2, and by decreasing the levels of nicotinamide (NAM). 49 Similarly, the genetic interventions that specifically decrease NADH levels increase life span, validating the model that NADH regulates yeast longevity in response to CR. 29 On the contrary, deletion of either SIR2 or NPT1 nullified the beneficial effect of CR. 30 In addition, NAD+ precursors and nicotinamide riboside (NR) have been shown to slow down aging and extend life span in yeast 28 and mammalian cells 20 and protect severed axons from degeneration in animal models of neurodegenerative diseases. 34 There is little information on the ways to increase NAD+ with commercially available natural products. Although NAD+ and NADP+ are recycled back and forth between oxidized (NAD+ and NADP+) and reduced forms (NADH, NADPH), cells require ongoing NAD+ synthesis because of the activity of NAD+-consuming enzymes, which are induced by stresses such as DNA damage and inflammation. 50,51 For example, tumor necrosis factor-α (TNF-α) and oxidative stress significantly reduce NAMPT and NAD+ levels in primary hepatocytes. 44 Tryptophan, nicotinic acid, nicotinamide, NMN, NR, and possibly nicotinic acid riboside are NAD+ precursors and can be utilized through distinct metabolic pathways to form NAD+ (Fig. 1 19,50 ). It was reported that nicotinamide is a better precursor of NAD+, while nicotinic acid is rapidly cleared by conversion to nicotinamide and excreted as nicotinuric acid when radiolabeled nicotinamide and nicotinic acid (niacin) were administered to mice. 19 NR supplementation in mammalian cells and mouse tissues increases NAD+ levels and activates SIRT1 and SIRT3, culminating in enhanced oxidative metabolism and protection against high-fat diet-induced metabolic abnormalities. 52 Mice fed NR in high doses in combination with their high-fat diets minimized their risk of getting diabetes, while not gaining weight. 52 Yoshino et al. 44 showed that NAMPT-mediated NAD+ biosynthesis is severely compromised by high-fat diet in metabolic organs of type 2 diabetes mice and that NMN can restore NAD+ levels and improve type 2 diabetes complications, partly through SIRT1 activation. Besides, mitochondria in muscles of elderly mice can be reversed to a youthful state after injections with NMN, thus raising NAD+ levels in old mice, restored mitochondrial function to that of a young mouse in a SIRT1-dependent manner. 53 Gomes et al. 53 proposed a model linking decreased NAD+ to loss of nuclear SIRT1 activity to stabilization of the hypoxia-associated transcription factor, hypoxia-inducible factor 1-alpha (HIF-1a). HIF-1a promotes a hypoxic-like (Warburg effect) state in the cell. Additionally, in a recent experiment, Khan et al. 54 treated mitochondrial myopathy mice with NR; NR efficiently delayed early- and late-stage disease progression by inducing mitochondrial biogenesis in skeletal muscle and brown adipose tissue.
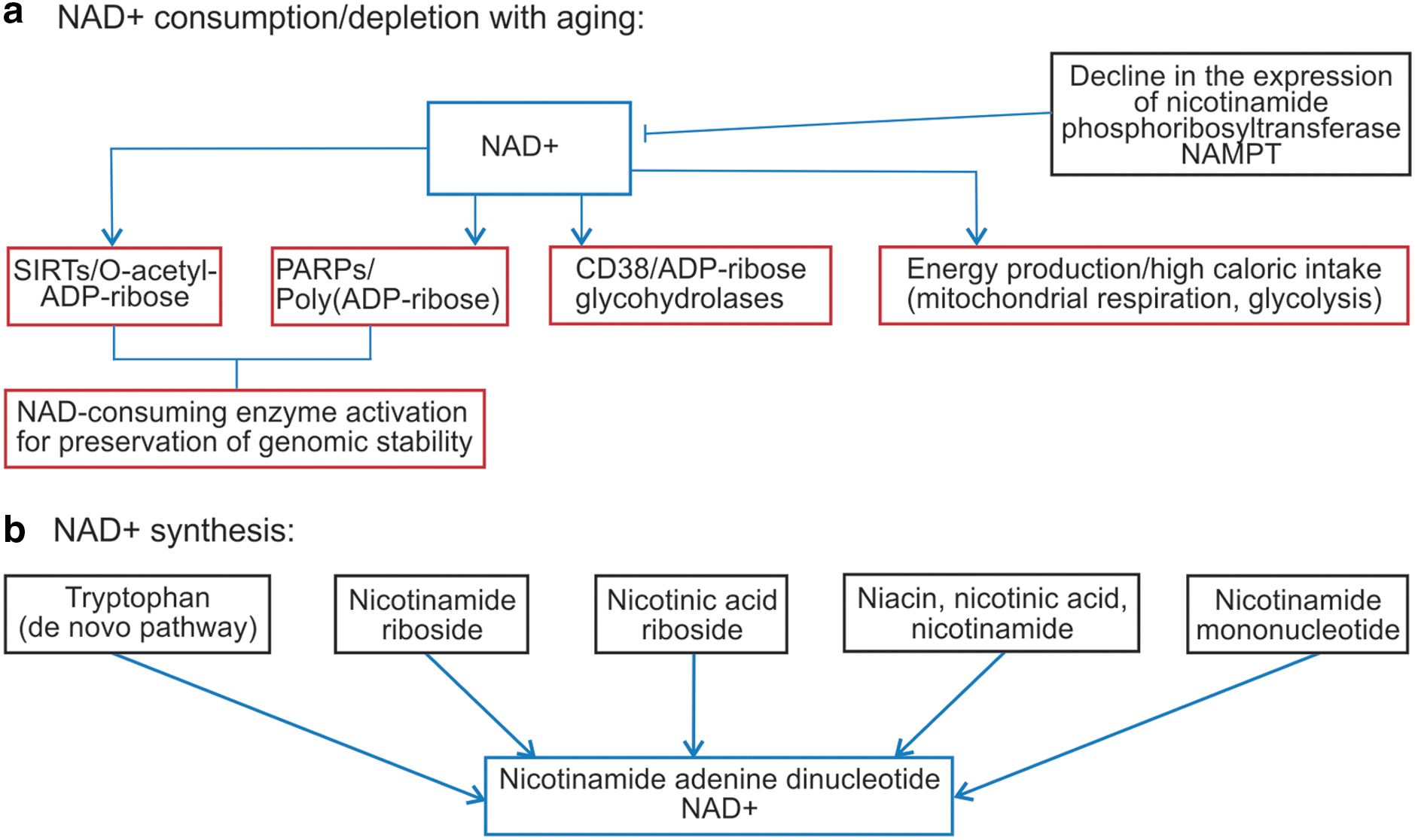
Pathways of NAD+ depletion
On the contrary, changes in NAD+ metabolism have been associated with several pathologies, including neurodegenerative diseases, cancer, cardiovascular disease, and normal aging. 34,55 The study of Braidy et al. 2 found a statistically significant decline in NAD+/NADH ratios and in intracellular NAD+ with age in the organs of rats. These changes in NADH occurred in parallel with an increase in lipid peroxidation and protein carbonyl formation and a decline in total antioxidant capacity of these organs. An age-dependent increase in DNA damage was also observed in these same organs. Decreased SIRT1 activity and increased acetylated p53 were observed in organ tissues in parallel with the drop in NAD+ and moderate overexpression of Sirt1 protein. The strong positive association observed between DNA damage, NAD+ depletion, and Sirt1 activity suggests that adequate NAD+ concentrations may be an important longevity assurance factor. 2 While Belenky et al. 56 observed that NR promotes Sir2 silencing and extends life span in yeast through Nrk and Urh1/Pnp1/Meu1 pathways to NAD+, Bitterman et al. 49 showed that physiological concentrations of nicotinamide noncompetitively inhibit both yeast Sir2 and human SIRT1 in vitro. Overexpression of nicotinamidase in Drosophila, which promotes the breakdown of nicotinamide, significantly increases median and maximal fly life span. 57
Therapies involved in maintaining high NAD+ levels may alleviate age-associated disorders and delay the aging process. 23,36 Beside supplementation with NAD+ precursors, 58 many studies have shown the protective effects of poly(ADP-ribose) polymerase (PARP) or CD38 inhibitors, for example, in ischemic heart and liver, diabetic kidney disease, and endotoxin-induced acute lung injury, most probably due to increased NAD+ bioavailability and the preservation of NAD+ levels. 2,59 –63 PARP inhibitors could be used also as anticancer agents 36,64 –67 by inhibiting DNA repair and increasing the potency of anticancer cytotoxic agents. Additionally, by increasing intracellular NADH levels, apoptosis can be induced. 68 –70
SIRT1/Sir2 and Poly(ADP-Ribose) Polymerase-1 Competition for NAD+
Sirtuins are NAD+-consuming deacetylases and PARP family members are also NAD+-consuming enzymes. 58 The activities of the PARP-1 enzyme, the predominant PARP activity in most tissues that is a major cellular NAD+ consumer, and SIRT1 are interrelated due to the competition for the same limiting intracellular NAD+ pool. 71 –74 NAD+ depletion may play a major role in the aging process by (1) limiting energy production, (2) DNA repair, and (3) genomic signaling. 34 The mammalian ADP-ribosyl cyclase CD38 could be a major regulator of NAD+ levels under physiological conditions, while (during severe DNA damage) PARP-1 could mediate intracellular NAD+ concentration. 75,76 Massudi et al. 34 commented that overactivation of PARP-1 has been reported in the brains of Alzheimer's disease patients as well as in those with diabetes, MPTP-caused Parkinson's disease (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), shock, and other conditions. SIRT1 translates alterations of NAD+ levels into transcriptional events. 77 By increasing cellular NAD+ levels, AMPK enhances SIRT1 activity, resulting in the deacetylation and modulation of the activity of downstream SIRT1 targets. Sirtuins are NAD+-dependent histone deacetylases/deacylases important for aging. This is especially true for the mammalian sirtuin 1 whose activity depends on the NAD+/NADH ratio. It seems that NAD+ can activate sirtuins and regulate the genes involved in mitochondrial function, metabolic adaptations, apoptosis, inflammation, and many aspects of the aging process. 72,78,79 Sirtuins have been implicated also in many degenerative diseases such as cancer, diabetes, obesity, and neurodegenerative diseases. 78
The DNA repair enzyme, PARP, also uses large amounts of intracellular NAD+ and is thereby in competition with sirtuins for the limited supply of NAD+. 58,80 Poly(ADP)-ribosylation is a DNA strand break-driven post-translational modification of nuclear proteins that is catalyzed by PARP-1, with NAD+ serving as a substrate and a key player in the immediate cellular response to ROS-induced DNA damage in eukaryotic cells. Cellular responses according to PARP activation intensity may vary (Fig. 2). Upon DNA damage, PARP activity in the cell is highly enhanced. Overactivation of PARP-1 following chronic oxidative stress and extensive DNA damage would lead to NAD+ depletion 81,82 and elevation in a by-product nicotinamide, therefore limiting SIRT1 activity by decreasing the bioavailability of this crucial coenzyme. 83 Deacetylation by Sir2 is dependent on high concentrations of NAD+ and inhibited by physiologic levels of nicotinamide. 24,84 The reduced effectiveness of sirtuins (SIRT1) can deacetylate tumor suppressor proteins such as p53, resulting in negative regulation of p53-mediated transcriptional activation. 85 Unlike PARP-1, the impact of PARP-2 on SIRT1 activity is not necessarily based on the changes in NAD+ content. The increase in SIRT1 expression observed in the PARP-2-deleted mice was due to PARP-2 acting as a direct negative regulator of the SIRT1 promoter. 80
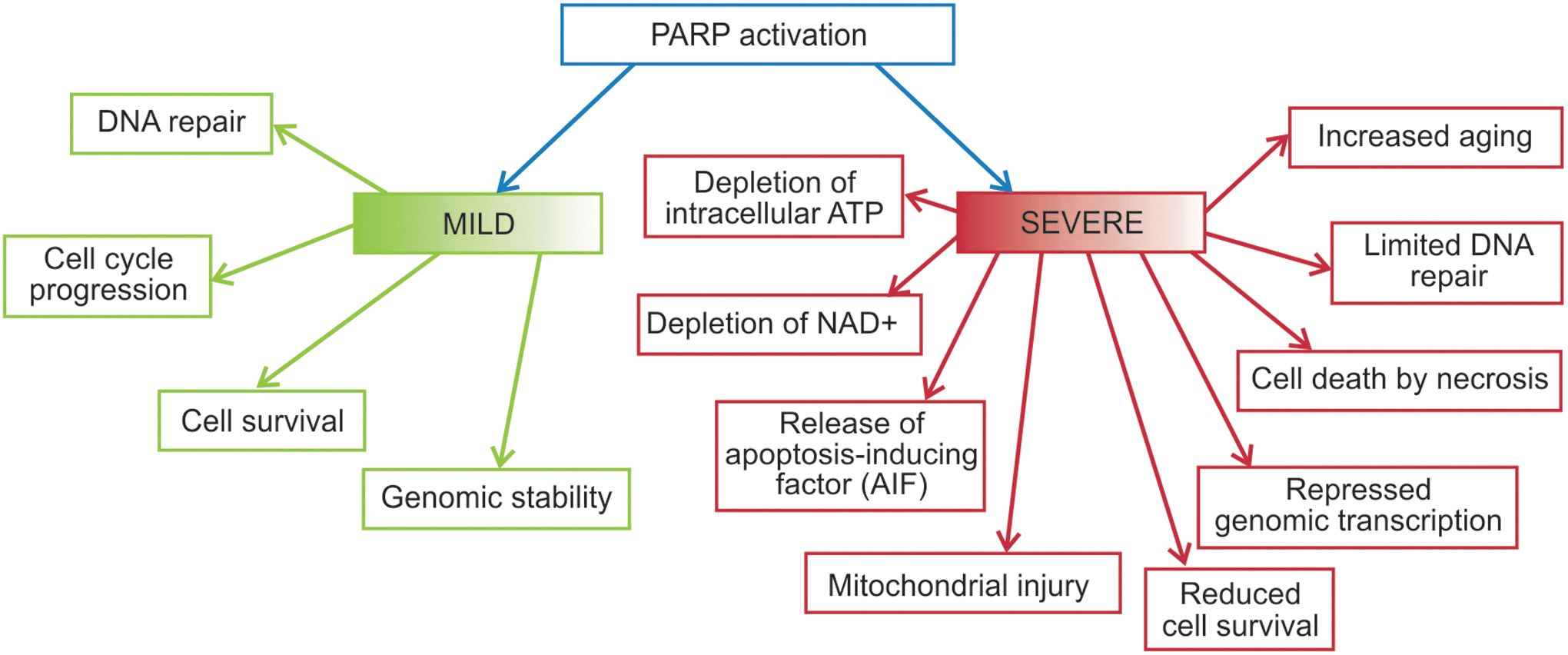
Intensity of PARP activation induces different cellular responses. PARP, poly(ADP-ribose) polymerase. Color images available online at
It seems that longevity is associated with a higher poly-ADP-ribosylation capacity as PARP is increased by 1.6-fold in centenarians 86 and PARP activity of 13 mammalian species correlates with a species-specific life span. 87 Moderately increased poly(ADP-ribosyl)ation capacity in long-lived species might slow down the rate of aging by preventing accumulation of DNA damage more efficiently compared with short-lived animals. 88
The Age-Dependent Mitochondrial Vicious Cycle and Aerobic Glycolysis
Mitochondrial dysfunction is highly tissue and context dependent, and it is perceived as a hallmark of aging; however, its causes are still debated. 53,89,90 Besides, whether mitochondrial dysfunction is a cause or a consequence of cellular impairment and the aging process, is a subject of intense debate. 89,90 Previous hypotheses speculated about free radical-induced oxidative stress as the main cause of mitochondrial inner membrane damage that creates a positive feedback loop. Excessive production of ROS generates mtDNA mutations, 91 –93 in turn leading to a defective respiratory chain and stimulation of glycolysis. 94 The defective respiratory chain generates even more ROS, ROS generate more damage to mtDNA, the reduced energy formation from oxidative phosphorylation (OXPHOS) further stimulates aerobic glycolysis, and a vicious cycle is generated. de Grey proposed that mtDNA mutations shift the intramitochondrial NAD+/NADH redox couple toward NADH, resulting in increased ROS production. 95 Although at a younger age very few cells suffer OXPHOS collapse resulting in increased glycolysis, 96 the problem increases with age when there is a metabolic shift from the use of mitochondrial energy toward reliance on glycolysis. 97
The Bioavailability of NAD Influences Mitochondrial Vicious Cycle and Aerobic Glycolysis
In this study, we introduce the role of the NAD+/NADH ratio to the mentioned vicious cycle as the bioavailability of NAD+ is the limiting factor for maximal oxidative capacity of mitochondria during OXPHOS as well as during glycolysis. Glycolytic cells generate reductive stress caused by increased NADH production during TCA cycle activity and glycolysis. Maintenance of the cytoplasmic NAD+/NADH ratio, by NADH reoxidation, is essential for continued glucose oxidation, ATP production, and normal cell functioning. 98 The obligate requirement for the oxidizing power of NAD+ to produce two ATP/glucose during glycolysis must be accompanied by a corresponding regeneration of NADH to NAD+. 97 Glycolytic cells recycle NADH through lactate dehydrogenase and plasma membrane electron transport (PMET)/plasma membrane oxidoreductase (PMOR). 99,100 Cells that generate most of their energy through OXPHOS recycle NADH mainly through mitochondrial electron transport. As NAD+ levels decline with age, mitochondrial function is impaired 56 and the DNA repair activity declines as well. 101 The increase of DNA damage with age 34 may therefore be the result of (1) an increased ROS generation and (2) a decline of DNA repair mechanisms and clearance affected by lower availability of NAD+ for sirtuins and PARPs. Several mitochondrial functions decline with age and the efficiency of removal of malfunctioning mitochondria also declines, 14 resulting in more mitochondria producing higher levels of superoxide, which is a precursor for other ROS and oxidative stress-induced damage. Mitochondria of older organisms are fewer in number, larger in size, and less efficient. 14,102 On the other hand, the vicious cycle theory, which states that free radical damage to mitochondrial DNA leads to mitochondria that produce more superoxide, has been questioned by some scientists since the most damaged mitochondria are degraded by phagocytosis, whereas the less defective mitochondria (which produce less ATP as well as less superoxide) remain to reproduce themselves. 103 However, the efficiency of lysosomes to consume malfunctioning mitochondria, that is, autophagic activity, declines with age, 104 resulting in more mitochondria producing higher levels of superoxide.
NAD+ Depletion Influences ATP Production, ROS Generation, and Antioxidative Defense
It seems that with advancing age, the NAD+ level (and NAMPT activity) declines and NADH level increases. 2,105 In this study, we propose an alternative explanation—dysfunctional mitochondria and increased glycolysis produce the state of reductive stress, where NADH is generated in excess and the amount of NAD+ is depleted. Depleted NAD+ influences mitochondrial ATP production, reductive stress, and ROS generation, as well as impaired antioxidative defense and DNA repair. For example, age-accelerated mitochondrial dysfunction was reported when cells lacked sufficient NAD+. 106,107 High levels of cytosolic NADH can additionally inhibit OXPHOS by promoting pyruvate to lactate conversion and reducing the permeability of the voltage-dependent anion channel in the mitochondrial outer membrane. 108 NAD+ is necessary for NADPH formation, which is required for glutathione and H2O2-inactivated catalase regeneration, and important in a thioredoxin antioxidant system. 76,109,110
OXPHOS and PMET/PMOR System can Regenerate NAD+
NAD+ can be regenerated through oxidation in the electron transport chain. Similarly, the PMOR system is able to accept electrons from intracellular NADH, thus regenerating NAD+ and export electrons out of the cell, as long as some compound is present in the extracellular medium that can then absorb them. The PMOR's physiological electron acceptor is unknown. Such an electron acceptor could be oxygen, 76,100 –113 thus yielding superoxide, which is further converted into other ROS. 103 PMET has a vital role in maintaining both plasma membrane and intracellular redox balances to support continued cell survival, function, and growth especially in fast proliferating cells. 99,114,115 Perturbation of key redox couples such as the NADH/NAD+ ratio can have profound effects on cells, resulting in apoptosis 114,115 and accelerated aging. For example, De Luca et al. 114 proposed that inhibition of PMET/PMOR through tumor-associated NADH oxidase (tNOX) results in increased levels of intracellular NADH and membrane ubiquinol, inhibiting sphingosine kinase and increasing membrane ceramide levels that may induce G1 arrest and apoptosis. Thus, ameliorating the NAD+/NADH ratio would influence the intensity of superoxide generation from the transfer of electrons to molecular oxygen at mitochondrial complexes I/III and from the plasma membrane redox system and can thus regulate the formation of reductive/oxidative stress. It is not just NAD+ breakdown reactions and biosynthetic (de novo and salvage) pathways that are important for adequate NAD+ intracellular concentration but also OXPHOS and PMOR systems that can regenerate NAD+ from NADH.
Conclusion
In the hypothesis presented, NAD+ was shown as an important factor related to aging at the cellular level, capable of reversing many of the age-related changes caused by decreased NAD+ levels. It is not the function of NAD+ as a cofactor in redox reactions that has the most important role in aging, but rather the role of NAD+ as a substrate for sirtuins and PARP activity. The beneficial anti-aging effects of NAD+ in humans have to be characterized as well as the form and amount of NAD+ supplementation that is most appropriate for human nutritional use. While we wait for the magic pill that would increase NAD+, CR and physical activity remain the best strategies for ameliorating NAD+ levels. The current knowledge on NAD+ regulation is based on work in cell cultures and model organisms. The proposed ideas are mostly limited to cellular aging and provide limited insights into the control of organismal aging. Declined aging and extended life span were shown in yeasts treated with NAD+ precursors. 34 Aging in humans is a much more complex process and caution is needed when extrapolating the knowledge from the studies in unicellular to multicellular organisms (e.g., NAD+ biosynthetic pathways differ in lower eukaryotes and invertebrates vs. mammals 43,116,117 ). Nevertheless, the NAD+ role in regulating longevity through the SIRT family seems to be evolutionary conserved from microorganisms to man. 22,78 Much of what we learned by using microorganisms as model systems to study aging will improve our understanding of these mechanisms in mammals and other higher eukaryotes. Further in-depth studies and human trials are needed before the modulation of NAD+ levels can be used to delay aging in humans.
Footnotes
Acknowledgment
The work was funded partially under the H2020-EU.4.a.WIDESPREAD-1–2014-Teaming ARTEMIDA Project 664536.
Author Disclosure Statement
There are no conflicts of interest. The work has not been published elsewhere, either completely, in part, or in another form. The manuscript has not been submitted to another journal and will not be published elsewhere. The article does not contain experiments using animals. The article does not contain human studies.