
Abstract
Reduced telomere length with increasing age in dividing cells has been implicated in contributing to the pathologies of human aging, which include cardiovascular and metabolic disorders, through induction of cellular senescence. Telomere shortening results from the absence of telomerase, an enzyme required to maintain telomere length. Telomerase reverse transcriptase (TERT), the protein subunit of telomerase, is expressed only transiently in a subset of adult somatic cells, which include stem cells and smooth muscle cells. A recent report from Xiong and colleagues demonstrates a pivotal role for the transcription co-factor peroxisome proliferator-activated receptor γ co-activator-1α (PGC-1α) in maintaining TERT expression and preventing vascular senescence and atherosclerosis in mice. Ablation of PGC-1α reduced TERT expression and increased DNA damage and reactive oxygen species (ROS), resulting in shortened telomeres and vascular senescence. In the ApoE−/− mouse model of atherosclerosis, forced expression of PGC-1α increased expression of TERT, extended telomeres, and reversed genomic DNA damage, vascular senescence, and the development of atherosclerotic plaques. Alpha lipoic acid (ALA) stimulated expression of PGC-1α and TERT and reversed DNA damage, vascular senescence, and atherosclerosis, similarly to ectopic expression of PGC-1α. ALA stimulated cyclic adenosine monophosphate (cAMP) signaling, which in turn activated the cAMP response element-binding protein (CREB), a co-factor for PGC-1α expression. The possibility that ALA might induce TERT to extend telomeres in human cells suggests that ALA may be useful in treating atherosclerosis and other aging-related diseases. However, further investigation is needed to identify whether ALA induces TERT in human cells, which cell types are susceptible, and whether such changes have clinical significance.
Introduction
T
Inactivation of TERT leading to pathologically shortened telomeres in mice has been shown to reduce mitochondrial biogenesis and the transcription co-activator factor peroxisome proliferator-activated receptor-γ co-activator-1α (PGC-1α) via induction of p53. p53 antagonizes PGC-1α expression, causing mitochondrial and metabolic dysfunction that can be rescued by ectopic expression of PGC-1α or TERT. 3 Thus, PGC-1α can be viewed as a suppressor of telomere-mediated pathology.
Deleting one copy of PGC-1α in an atherosclerotic knockout apolipoprotein E (ApoE−/−) mouse strain reduces expression of PGC-1α by 50%. This effect results in more severe atherosclerosis, as well as vascular senescence characterized by reduced TERT, sirtuin 1 (SIRT1), and catalase, as well as increased p53 expression. Thus PGC-1α may play a key role in preventing vascular senescence and atherosclerosis. 4
TERT and PGC-1α Cooperate to Protect Cells from DNA Damage and Mitochondrial Dysfunction
In a recent study, Xiong et al. 7 extended previous work to identify mechanisms by which PGC-1α and TERT cooperate to protect cells from mitochondrial dysfunction, DNA damage, and vascular senescence beyond stimulating SIRT1 transcription. 4 This group 7 discovered that the ApoE−/− mouse model of atherosclerosis, in which old mice develop atherosclerotic plaques similar to human atherosclerosis, can be augmented by knocking out both copies of the gene encoding PCG-1α. They constructed a new double knockout mouse model (ApoE−/− PGC-1α−/−), in which old (18- to 20-month-old) mice fed a standard diet develop more severe atherosclerosis and increased vascular senescence than the ApoE−/− mice, suggesting that PGC-1α helps protect the vasculature from development of atherosclerosis. 7
On the basis of previous work in which TERT deficiency reduced PGC-1α expression, 3 Xiong et al. hypothesized that levels of TERT, which is known to be expressed in vascular smooth muscle cells, would be reduced. Indeed, western blot analysis showed reduced TERT expression, whereas telomerase assays showed a consistent 50% reduction of activity in the aortas of middle-aged mice. In old mice, a further 50% reduction in TERT expression and activity was observed. At the same time, vascular senescence, as measured by age-associated β-galactosidase (AABG) expression increased inversely with decreased TERT expression. As expected, telomere length was shortened significantly to an average of 6 kb using Southern blot analysis in primary mouse aortic smooth muscle cells (MASMs). 7 The authors do not mention that “short” 6-kb mouse telomeres are still twice as long as average adult human telomeres. However, forced expression of a constitutively active PGC-1α or TERT using an adenovirus gene therapy vector reduced expression of AABG to normal levels, whereas at the same time restoring telomere length to 10–12 kb, similar to normal control cells.
Although these results suggest that telomere length may matter, they beg the question of how such “short” telomeres in mice, that are actually relatively long in humans, behave so differently across species. The answer may lie in the extent of telomere-associated DNA damage. Xiong et al. hypothesized that increased DNA damage would result from increased reactive oxygen species (ROS), which are associated with the atherosclerotic phenotype and PCG-1α deficiency. They observed moderately increased ROS in PGC−/− aortas and mouse aortic smooth muscle MASMs, as well as increased levels of 8-hydroxydeoxyguanosine (8-HG), a characteristic of oxidatively damaged DNA. Moreover, comet assays detected single- and double-strand breaks in DNA. Consistent with data on restored telomere length, ectopic expression of either TERT or PGC-1α restored 8-HG levels to normal. 7 But what about possible telomere DNA damage? Xiong et al. suggest that these data support the idea that the absence of PGC-1α causes TERT down-regulation, which in turn causes DNA damage and telomere shortening/dysfunction.
Increased ROS was hypothesized to cause the observed DNA damage. To test this idea, Xiong et al. treated cells with N-acetylcysteine (NAC), a strong anti-oxidant, to reduce ROS levels. NAC (5 mM) decreased levels of ROS in PCG-1α−/− ApoE−/− MASMs in culture to that of wild-type MASM controls. As expected, there was a beneficial effect of NAC, with about 50% reduction in the senescence biomarker AABG, a 2.5-fold induction of TERT/telomerase activity, and a three-fold reduction of 8-HG. However, even after NAC treatment, there was still 50% more AABG, five-fold less TERT, and two-fold less damage as assessed by the comet assay, suggesting that the defects associated with the absence of PGC-1α were due to more than just increased ROS and oxidative DNA damage. Tellingly, telomere length increased moderately with NAC treatment, but still was not restored to wild-type levels. This raised the possibility of telomere dysfunction and associated DNA damage.
To investigate possible telomere DNA damage, cells were stained immunofluorescently with antibodies to gamma-H2AX, a histone that accumulates at double-stranded breaks. At the same time cells were stained with fluorescently labeled, telomere-specific peptide nucleic acid probes to determine the location of the telomeres. Gamma-H2AX staining showed significant evidence of DNA damage in PCG-1α−/− ApoE−/− MASMs (seven-fold more) compared with wild-type cells, split almost evenly between telomeric and non-telomeric foci. Treatment with NAC reduced non-telomeric damage to within two-fold of normal levels, but only reduced DNA damage at telomeric foci by about 10%, so that telomere DNA damage remained at least four-fold greater than controls. They concluded that oxidative stress associated with loss of PGC-1α strongly contributes to genomic DNA damage and moderately contributes to telomere shortening and telomeric DNA damage.
But clearly there is some other factor causing telomeric DNA damage, most likely reduced TERT. Xiong et al. hypothesize that reduced TERT expression is not only due to ROS, but to loss of PGC-1α directly. 7 This makes sense and may explain the species difference between mice and humans alluded to earlier: TERT not lengthens telomeres, but also stabilizes them to confer protection from DNA damage. Also, it has been reported that telomerase can protect mitochondrial DNA from damage, 8 which may also play a role in how PGC-1α and TERT reverse senescence, although mitochondrial DNA damage was not assayed by Xiong and colleagues. So the absolute length of telomeres is only one factor. This idea could be tested by making cells conditional for TERT expression.
To test the hypothesis that PCG-1α directly regulates TERT transcription, Xiong et al. analyzed the TERT promoter and found evolutionarily-conserved DNA-binding elements of the transcription factors (TFs) peroxisome proliferator-activated receptor-γ (PPARγ) as well as FoxO1, Nrf-2, p53, cAMP response element-binding protein (CREB), estrogen receptor-α/estrogen-related receptor-α (ER-α/ERR-α), and Ying Yang 1 (YY-1). PGC-1α control the expression of all these TFs. 9 In support of this idea, over-expression of constitutively active PGC-1α in rat aortic smooth muscle cells (RASMs) increased TERT in a dose-dependent manner. 7 Surprisingly, similar evidence from MASMs or human aortic smooth muscle cells was not provided. Moreover, multiple pathways with anti-inflammatory and pro-mitochondrial activity were stimulated: Nrf-1, Nrf-2, mitochondrial transcription factor A (TFAM), SIRT1, heme oxygenase-1 (HO-1), and manganese superoxide dismutase (MnSOD). Of special interest were Nrf-2 and HO-1, which are important for anti-oxidant-/electrophile-responsive element (ARE/ERE) adaptive electrophilic responses. 10
Xiong et al. postulated that alpha-lipoic acid (ALA), a compound that previously was observed to reduced atherosclerosis and increase PGC-1α expression in mice, 11 would increase TERT and ARE/ERE signaling to decrease DNA damage and improve telomere function. In normal RASMs, ALA induces PGC-1α about three-fold and TERT about two-fold, confirming their hypothesis. Moreover, treatment of young ApoE−/− mice with 3–10 mg/kg ALA increases telomerase activity 2.5-fold, increases TERT and ARE/ERE signaling significantly over background, and reduces p53, which is known to reduce TERT expression levels by two-fold in a dose- and time-dependent manner. To confirm that PGC-1α is required, Xiong et al. repeated this experiment on PGC-1α−/− MASMs and found no stimulation of TERT or ARE/ERE signaling. 7
Xiong et al. next tested the hypothesis that CREB mediates the effects of ALA on PGC-1α. CREB is known to enable PGC-1α transcription, 12 and its down-regulation is associated with atherosclerosis. 13 ALA strongly stimulates cAMP in mice aortas and smooth muscle cells for 2 days after treatment and activates phosphorylation at Ser133 of CREB; however, CREB expression levels were unchanged, suggesting that ALA may stimulate PGC-1α through increased cAMP via CREB. 7
A high-fat diet (HFD) is known to be pro-atherosclerotic in ApoE−/− mice. 14 Xiong et al. observed that ApoE−/− mice fed a HFD for 2–4 weeks exhibited reduced PGC-1α, SIRT1, and TERT/telomerase activity, but increased p53 levels. Treatment of youthful HFD ApoE−/− mice with injected ALA for 7 days, completely blocked the reduction of PGC-1α, SIRT1, TERT/telomerase activity, and the increase in p53, while reducing DNA damage to background levels. Vascular aging, as assessed by AABG and atherosclerotic plaques, was reduced to below background levels after ALA treatment. 7
It should be noted that these studies were performed not only in rodents but also mostly in genetically manipulated mouse models, which are not physiological, and that careful biochemical studies in normal animals and humans are needed to extend the applicability of this work. Xiong et al. suggest that these data are consistent with a model wherein TERT and PGC-1α form an inhibitory positive feedback loop, whereby loss of either one promotes the loss of the other (Fig. 1). It is intriguing that the DNA regulatory elements that control PGC-1α are conserved in humans and suggest that these results, or at least this control loop, may translate to humans, although confirmatory experiments on human cells and subsequently in humans are required.
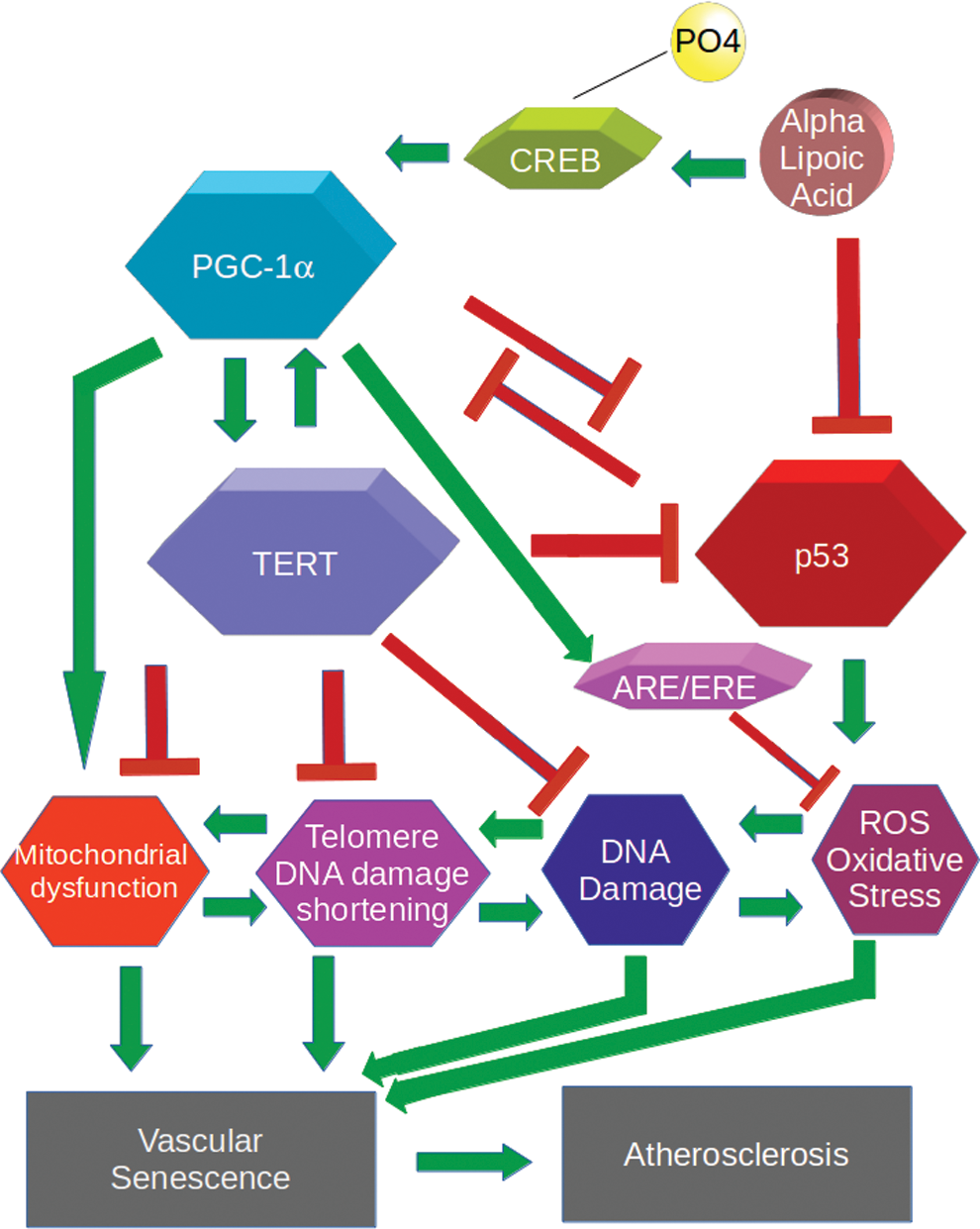
Peroxisome proliferator-activated receptor-γ co-activator-1α (PGC-1α) stimulates telomerase reverse transcriptase (TERT) to mitigate telomeric and genomic DNA damage, reactive oxygen species (ROS), mitochondrial dysfunction, and senescence in vascular smooth muscle cells. PGC-1α stimulates transcription of TERT, which in turn reduces genomic and telomeric DNA damage and mitochondrial dysfunction, reversing vascular senescence and atherosclerosis. PGC-1α stimulates multiple anti-inflammatory pathways (not shown), including anti-oxidant-/electrophile-responsive element (ARE/ERE), which promote mitochondrial function. Mitigation of DNA damage and ROS lower p53 levels to allow increases in TERT and PGC-1α expression. ALA stimulates PGC-1α transcription via phosphorylation of the transcription factor cAMP response element-binding protein (CREB), to stimulate TERT expression and relieve vascular senescence and atherosclerosis. Color images available online at
Medical Implications
There is great interest in stimulating telomerase expression to alleviate shortened telomeres that are associated with aging, especially in lymphocytes. 15 Because ALA augments telomerase in rat MASMs and mouse aortas, its ability to stimulate TERT and telomerase activity in corresponding human cells and especially lymphocytes should be investigated. Should ALA increase telomere length in humans, it may prove to be a relatively safe way to control telomere length. ALA is available over the counter as an oral supplement and has been used in clinical trials for diabetes and other ailments. In Germany, injected ALA is a prescription medicine to treat type 2 diabetes. 16,17 On the other hand, clinical trials with ALA have failed to produce data that are consistent with the results reported by Xiong et al., although specific end points consistent with this report have not been explored. 18 –21
Preventing replicative senescence and/or telomere DNA damage during aging could be of benefit, because elimination of some senescent cells has been reported to rejuvenate some tissues, which presumably are affected by the secreted associated senescent phenotype (SASP). 20,21 If ALA can actually stimulate TERT in a wide variety of human cells, then it may have serious benefit for maintaining quality of life during aging. However, supplementation with ALA in adult mice did not lead to increases in health span or life span, 23 casting some indirect doubt on its capability to affect TERT in a large variety of cells, as ectopic TERT expression in normal mice promotes health span. 24 It will be interesting to elucidate how permissive most adult somatic cells that do not normally express TERT are to TERT induction. Furthermore, it will be useful to understand the mechanism that underlies how ALA stimulates PGC-1α. If ALA stimulation of cAMP activates CREB, which in turn promotes the expression of PGC-1α (Fig. 1), then how does ALA stimulate cAMP? At least one report suggests that ALA stimulates cAMP through G-coupled membrane receptors. 25 If true, how universal are cAMP stimulation, CREB activation, and induction of PGC-1α and TERT?
It is informative that increased DNA damage and senescence are associated with reduced expression of a master transcription factor like PGC-1α through its interaction with TERT. That PGC-1α can suppress senescence and DNA damage, and in some cases reverses these phenotypes, suggests that therapies based on stimulating PGC-1α could have benefit in aging.
However, it is quite premature to believe that enhanced expression of PGC-1α will increase human longevity. To date, studies in humans have found no correlation of longevity with PGC-1α levels or with mitochondrial biogenesis. 26 In fact, data suggest that mitochondrial biogenesis in “successful” old age is reduced and that the transcription factor YY1 has a negative correlation with longevity. 26 Moreover, no single-nucleotide polymorphisms (SNPs) have been reported in the PGC-1α gene that associate with longevity, although several polymorphisms were associated with Parkinson's disease in one study, 27 but were not confirmed in another. 28
Conclusion
PGC-1α expression prevents vascular senescence in a mouse model of atherosclerosis by maintaining TERT levels through an inhibitory positive feedback loop. ALA is able to induce PGC-1α, TERT, and other pro-mitochondrial signaling pathways, possibly by activation of CREB via phosphorylation. ALA may be a general-purpose treatment to increase telomere length, although this idea requires further investigation.
Footnotes
Author Disclosure Statement
No competing financial interests exist.