
Abstract
Prolonged physiological stresses, including abnormal pH and temperature, are deleterious. However, human hepatic progenitors have been shown to be quite tolerant of temporary temperature stress such as in cold ischemia. We aimed at identifying how various stresses affect liver progenitors, and at determining whether distinct effects exist on different progenitor cells of the human liver. Total fetal liver cells were exposed to low (25°C), normal (37°C), or high (40°C) temperatures, or low (6.76), normal (7.35), or high (7.88) pH in vitro. Culture at 25°C increased cell numbers and percentages of proliferation marker Ki67+ total cells. In total cell cultures, percentages of CD326+ hepatic progenitors co-expressing DLK1 (delta-like 1 homolog), SSEA4, or CD90 increased, as well as proliferation of SSEA4+ and CD235a+ progenitors. Analyses of presorted hepatic progenitors revealed that culture at 25°C increased cell numbers of CD326+ hepatic stem/progenitor cells but not DLK+ hepatoblasts. The expression of several mesenchymal genes was reduced, and distinct hepatic stem/progenitor cell colonies emerged. At 40°C, numbers of adherent hepatic cells decreased but those of hematopoietic nonadherent cells increased. High pH did not cause major effects. Acidic pH resulted in decreased total cell numbers and affected hematopoietic cells. Percentages of DLK1+ hepatoblasts were increased, but those of hematopoietic mature CD45+ cells were decreased. In particular, proliferation of adherent hepatic CD326+, SSEA4+ progenitors, and hematopoietic CD45+ cells and CD235a+ erythroblasts was reduced. Conclusively, our data indicate that low-temperature stress stimulates hepatic progenitor and erythroblast proliferation, whereas acidic pH promotes hepatic maturation and reduces hematopoietic cells.
Introduction
P
Surprisingly, there are not much data available on the effects of stresses on stem/progenitor cell types, yet it seems reasonable to speculate that an inductive response is triggered in stem cells by stresses, also to compensate for a potential loss of cells. From the adult liver, the isolation and culture of viable human hepatic stem/progenitor cells has been shown to be possible after up to 7 days of cold (4°C) ischemia. 7,8 Mild thermal stress of 41°C, but not increased thermal stress of 42.5°C, has been demonstrated to be beneficial on the healing capacity of wounds and on angiogenesis of human umbilical vein cells in vitro. 9 Similarly, mildly increased temperatures of 39°C induced proliferation and mineralization of human bone marrow mesenchymal stromal cells but caused growth arrest in osteosarcoma (Mg-63) cells. 10 Human bone marrow mesenchymal stem cells, subjected to various pHs ranging from 6.3 to 8.5 in culture, revealed the highest proliferation rates at alkaline pH8.0. 11 An acidic pH (below 6.0) has been shown to induce neural conversion of animal caps in classical experiments with salamander. 12
We aimed at analyzing the effects of stresses (i.e., low and high temperature as well as pH) on the various stem/progenitor cell populations of the fetal liver. The human fetal liver comprises various cell types of different embryonic lineage origin and is the main site of hematopoiesis during fetal development. To determine the effects on specific stem/progenitor cell types in cultures of total fetal liver cells, we employed fluorescence activated cell sorting (FACS) and used multiple cell surface markers for co-expression studies, in combination with nuclear Ki67 for proliferation analyses. We also employed magnetic activated cell sorting (MACS) to investigate effects on separated, sorted hepatic progenitors, that is, hepatic stem progenitor/cells and hepatoblasts.
With a few exceptions, most surface markers are not unique to one cell type and overlaps exist. Hepatic stem/progenitor cells can be detected by their expression of CD326. Different developmental stages of human hepatic stem/progenitors can be distinguished and have been described by others 13,14 and us. 15,16 In cell culture, hepatic cell types are adherent, whereas most hematopoietic cell types are nonadherent in suspension. The early, mesendodermal multipotent liver stem/progenitor cell is positive for CD326 and CD90; the endodermal hepatic stem/progenitor cell is positive for CD326 but negative for CD90 and DLK1; and its progeny, the hepatoblast, is positive for DLK1 but mostly negative for CD326. Hematopoietic cells include diverse mature nonadherent and immature adherent cell types, for example, granulocytes, lymphocytes, and basophils, all of which can be detected by their expression of CD45, with the exception of erythrocytes and their progenitors that are CD45−. Both the mature, nonadherent, enucleated erythrocytes, and the immature, adherent, nucleated erythroblasts express CD235a. Hematopoietic stem cells are CD34+, but CD34 expression also has been reported in other progenitor cell types. Several mesenchymal cell types and progenitors commonly express CD90. SSEA4 expression can be detected in various early stem cells of hepatic and hematopoietic lineages. Endothelial cells can be characterized by their expression of CD31. In addition to cell population characterizations in FACS, we investigated the effects of stresses on cell viability, expression of various lineage- and cell type-specific genes, cell number and relative size, as well as hepatic colony formation.
Materials and Methods
Liver cell isolation
The procedure for isolation of human fetal liver cells was previously described 15 and performed with slight modifications. Human fetal livers of 15–20 weeks of gestational age were obtained as anatomical gifts provided by the Allegheny Reproductive Health Center, Pittsburgh, PA. Organs were retrieved from abortions, after informed consent of the donor and approval by the local Institutional Review Board (approval #PRO12020516). Liver tissues were placed in Petri dishes that included digestion medium, and tissues were minced with scalpels. The digestion medium contained 0.6 mg/mL collagenase type IV, 120 U/mL DNase, 1% fatty-acid-free bovine serum albumin (BSA), 30 nM selenium (all Sigma-Aldrich, St. Louis, MO), and antibiotic-antimycotic mix in RPMI 1640 medium (Life Technologies, Carlsbad, CA). The tissue suspension was transferred to a Falcon tube and filled up to 50 mL with digestion medium. The tube was incubated at 37°C and agitated every 5 minutes. After 30 minutes, about 45 mL supernatant of the suspension (containing single cells) was transferred to a new Falcon tube; to the remaining 5 mL (containing still undigested tissue fractions) 45 mL of fresh digestion medium was added, followed by an additional incubation at 37°C for 30 minutes with agitation every 5 minutes. Cell suspensions were washed two times with RPMI 1640 medium (containing 1% fatty-acid-free BSA, 30 nM selenium, and antibiotic-antimycotic mix) by centrifugation at 300 g, and subsequently filtered through 40 μm pore-size cell strainers (Becton Dickinson, Bedford, MA).
Cell culture
Freshly isolated fetal liver cells were re-suspended in enriched RPMI1640 medium, 15 which was supplemented with 5% fetal bovine serum, 0.1% fatty acid-free BSA, 30 nM selenium, 540 μg/mL niacinamide, 5 ng/mL insulin, 10 ng/mL transferrin, free fatty acid mixture (2.36 μM palmitic acid, 0.21 μM palmitoleic acid, 0.88 μM stearic acid, 1.02 μM oleic acid, 2.71 μM linoleic acid, 0.43 μM linolenic acid), 0.1 μM hydrocortisone, 50 μM β-mercaptoethanol (all Sigma-Aldrich), 2 mM Glutamax, and antibiotic-antimycotic mix (Life Technologies). RPMI1640 medium contained 200 mg/mL sodium bicarbonate buffer to give a physiological pH of about 7.35 at 37°C when in a 5% carbon dioxide atmosphere.
For cell culture, culture flasks or chamber slides (for microscopy) were coated with a thin layer of rat-tail tendon collagen-1 (Becton Dickinson) at a concentration of 5 μg per cm2. Total fetal liver cells were seeded at a density of 100,000 total live cells (including red blood cells) per cm2. To allow for attachment, cells were cultured for 24 hours at 37°C in a 5% carbon dioxide atmosphere. After this initial attachment phase, cell culture medium was changed to enriched RPMI1640, as described earlier, but without serum. Cells were subject to pH or temperature stresses for additional 48 hours, or 6 days for some experiments, after which they were harvested for subsequent analyses, resulting in a total culture length of 3 or 7 days, respectively. Stresses of low or high pH were achieved by adjusting carbon dioxide percentages. All pHs of media were verified at least three times by using a pH meter (AccumetBasic AB15; Thermo Fisher Scientific, Waltham, MA). Controls were cultured at 37°C and 5% carbon dioxide having a physiological pH7.35 ± 0.01 (average ± standard deviation). Low pH stress cultures were kept at 37°C and 20% carbon dioxide, resulting in pH6.76 ± 0.01. High pH stress cultures were maintained at 37°C and 0.5% carbon dioxide, giving a pH7.88 ± 0.02. For temperature stress, cultures were kept at 5% carbon dioxide and 25°C for low-, and 40°C for high-temperature stress, respectively. As the pKa of sodium bicarbonate is slightly temperature dependent, we also verified the physiological pH of the media at different temperatures having 5% carbon dioxide. The pH at both temperatures remained physiological, with pH7.37 ± 0.04 at 25°C and pH7.32 ± 0.15 at 40°C.
Cell numbers and viability
The total cell numbers and viabilities of freshly isolated cells as well as cultured cells were determined by using a Neubauer chamber and trypan blue (Life Technologies) exclusion in phase-contrast microscopy (Zeiss Invertoskop C; Carl Zeiss, Jena, Germany).
Indirect cell viability analysis
The QuantiChrom Lactate Dehydrogenase (LDH) Kit (BioAssay Systems, Hayward, CA) was used according to the manufacturer's instructions to determine indirectly cell viability from cell culture medium samples. The enzyme LDH is released only from disintegrating cells, and thus is commonly used to indirectly determine cell viability. Culture medium samples were collected from 3 to 7 day cultures of 25°C and 37°C. Negative culture controls included wells with medium but without cells. Negative assay control included water, and positive assay control included calibrator of the kit. Medium samples were collected and frozen immediately and kept at −20°C until they were assayed. Assays were performed in clear flat-bottom 96-well plates (Corning Costar, Corning, NY). Into separate wells, 200 μL of water and calibrator were transferred. Ten microliters of each sample was transferred into separate wells, and 190 μL of Working Reagent was added and mixed. The optical density at 565 nm was read immediately and after 25 minutes, by using a Synergy H1 Hybrid multi-mode microplate reader and Gen5 data analysis software (BioTek, Winooski, VT). LDH activity was calculated as IU/L.
Gene expression analyses
The effect of stresses on cell type-specific gene expression was studied. Freshly isolated cells as well as cells subject to stresses for 48 hours in culture were lysed directly with RLT-buffer, and RNA was isolated by using shredder- and isolation columns (RNAeasy-mini kit; Qiagen, Valencia, CA), including DNA digestion by DNase treatment on columns. Concentrations of nucleic acids were determined fluorometrically by using Quant-iT Assay kits and Qubit fluorometer (Invitrogen). RNA was reverse transcribed to cDNA with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA). Gene expression was analyzed by real-time polymerase chain reaction by using the StepOnePlus system and software, and predesigned TaqMan probe and primer assay mixes (listed in Supplementary Table S1; Supplementary Data are available online at
Beta-actin served as a housekeeping gene for internal normalization. TaqMan assay mixes were: ACTA1: actin, alpha 1, skeletal muscle (alpha smooth muscle actin); ACTA2: actin, alpha 2, smooth muscle, aorta; AFP: alpha-fetoprotein; ALB: albumin; CASP3: caspase 3, apoptosis-related cysteine peptidase; CCNE1: Cyclin E1; CD34: CD34 molecule (cluster of differentiation 34); CIRBP: cold inducible RNA binding protein; CYP3A7: cytochrome P450, family 3, subfamily A, polypeptide 7; DLK1: delta-like 1 homolog (Drosophila); EPCAM: epithelial cell adhesion molecule (CD326); HNF4A: hepatocyte nuclear factor 4, alpha; HSPA1A: heat shock protein family A member 1A; KRT19: keratin 19, type 1 (cytokeratin 19); LGR5: leucine-rich repeat containing G protein–coupled receptor 5; MKI67: marker of proliferation Ki-67; PECAM1: platelet/endothelial cell adhesion molecule 1 (CD31); POU5F1: POU class 5 homeobox 1 (Oct4); PTPRC: protein tyrosine phosphatase, receptor type, C (CD45); THY1: Thy-1 cell surface antigen (CD90); TNF: tumor necrosis factor (TNF alpha); VWF: von Willebrand factor.
Expression was quantified by using the ddCt method. Freshly isolated cells served as positive controls; no template (water) was used as negative control. Each biological sample was run in triplicate. Cultures at control conditions (regular temperature of 37°C or pH7.35, respectively) were used as reference (set as 100%). Data were calculated from n = 5 biological repeats.
Immunocytochemistry
Cultured cells were fixed with 4% para-formaldehyde (Sigma-Aldrich) for 1 hour and washed three times with phosphate-buffered saline (PBS). Unspecific binding was blocked by incubation with 10% goat serum and 1% FcR-block (Miltenyi Biotec, Bergisch Gladbach, Germany) in PBS for 1 hour at room temperature (RT). Cells were incubated with primary antibodies (in 10% goat serum in PBS) for 1 hour at RT. Antibodies (Supplementary Table S2) were rabbit IgG anti-human alpha-fetoprotein (DakoCytomation, Glostrup, Denmark) and mouse IgG1 anti-human cytokeratin 19 (Thermo Fisher Scientific). Cells were washed three times with PBS and incubated with secondary antibodies (in 10% goat serum in PBS) for 1 hour at RT. Secondary antibodies were Alexa Fluor 568 goat anti-rabbit IgG and Alexa Fluor 488 goat anti-mouse IgG1, and nuclei of cells were stained with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific). Cells were washed three times, embedded in Aquamount (Polysciences, Warrington, PA), and cover-slipped. Images were acquired with a Nikon Eclipse TE300 microscope (Tokyo, Japan) equipped with a ProgRes MF camera and software (Jenoptik, Jena, Germany). Images were assembled in Adobe Photoshop CS3 Extended version 10.0.1 software (Adobe Systems, San Jose, CA).
Flow cytometry
The percentages and proliferation of different liver cell types were determined by co-expression analyses of cell type-specific surface markers and nuclear proliferation marker Ki67 by using fluorescence-activated cell sorting. Freshly isolated cells or cells in culture after applying stresses were examined. Nonadherent and adherent cells were analyzed separately. Nonadherent cells in medium were collected; adherent monolayers were washed twice with PBS (without calcium and magnesium) and detached by incubation with trypsin/EDTA (Life Technologies); the enzymatic action was stopped by addition of serum-containing medium. Cell pellets were re-suspended in cold buffer (0.5% BSA [Sigma-Aldrich], 2 mM di-sodium EDTA [Sigma-Aldrich] in PBS without calcium and magnesium [Life Technologies]), including 10% human FcR blocking reagent (Miltenyi Biotec). For cell surface marker staining, cells were incubated with antibodies or their respective isotype controls (Becton Dickinson) for 15 minutes at 4°C in the dark (Supplementary Table S2 for a list of antibodies and isotypes used). Cells were washed with buffer. For co-staining of the nuclear proliferation marker Ki67, loosened cell pellets were re-suspended in ice-cold 70% ethanol, including 1% fetal bovine serum. Cells were fixed and permeabilized for at least 2 hours at −20°C. After washing twice with buffer, cells were incubated with Ki67 antibody or isotype control for 30 minutes at RT in the dark. Cells were washed and analyzed with FACSAria II flow cytometer (Becton Dickinson) and FlowJo software version 9.5.2 (Tree Star, Ashland, OR). An initial forward versus side scatter gate was applied to exclude cell debris and doublets; subsequently, the relative cell sizes were compared in forward scatter histograms. Compensation beads (Becton Dickinson) were used to compensate for fluorochrome spectral overlap. Negative controls included nonstained cells and isotype control-stained cells.
Magnetic activated cell sorting
To determine whether the observed increase of total cell numbers and proliferation at low-temperature culture specifically affects hepatic stem/progenitor populations differently, we sought to investigate cultures of either only hepatic stem/progenitor cells or hepatoblasts. Hepatic stem/progenitor cells and hepatoblasts were isolated from total human fetal liver cell suspensions by using selection for CD326 and DLK1 positivity, respectively, by MACS according to the manufacturer's instructions (Miltenyi Biotec).
Freshly isolated total liver cell suspensions were washed and re-suspended in washing buffer (PBS without Ca and Mg, with 2 mM ethylenediaminetetraacetic acid (EDTA) and 0.5% BSA, pH7.2 [all Sigma-Aldrich]). Suspensions were incubated in washing buffer with 10% FcR-block (Miltenyi Biotec) to prevent unspecific receptor-mediated antibody binding and 0.1 mg/mL DNase (Sigma-Aldrich) to prevent cell aggregation. For selection of CD326+ hepatic stem cells, anti-CD326 magnetic beads (Miltenyi Biotec) were added to suspensions. Cells were incubated at 4°C for 30 minutes, washed, and re-suspended. For selection of DLK1+ hepatoblasts, suspensions were incubated first with DLK1-APC antibody (Novus Biologicals, Littleton, CO) at 4°C for 15 minutes, washed, re-suspended, and subsequently incubated with anti-APC magnetic beads at 4°C for 30 minutes. Bead-labeled cells were separated on LS-columns by using a Midi-magnet (Miltenyi Biotec) according to the manufacturer's instructions, and cell numbers and viabilities were determined as described. Sorted cells were seeded at 1000 cells per cm2 and cultured as described for total fetal liver cells.
Statistics
Data are given as means from n biological repeats ± standard deviation. Student's t-test was used to analyze statistically significant differences. Asterisks indicate p-values: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Results
Freshly isolated human fetal liver cells had high viabilities of 96.0% ± 3.8% (Table 1) as determined by trypan blue exclusion assay from 10 specimens. The average yield of total live cells per liver was 1,293,050,000 ± 699,604,668. Variations in the total yield occurred based on the gestational age and integrity of the specimen (due to the nature of the donation retrieval procedure, specimens are usually not obtained as a complete, whole organ). Based on flow cytometric analyses of 10 livers, the freshly isolated total liver cell suspensions contained 54.1% ± 15.0% CD326+ hepatic stem/progenitor cells and 5.9% ± 4.2%, DLK1+ hepatoblasts. Hematopoietic cell types included 26.6% ± 36.5% CD235a+ erythroblasts/-cytes, 1.9% ± 0.6% CD34+ hematopoietic progenitors, and 11.5% ± 3.4% CD45+ cells. Also, 30.1% ± 12.0% SSEA4+, 0.8% ± 0.8% CD90+, and 5.6% ± 1.2% CD31+ cells could be detected.
Human fetal liver cells were initially seeded at densities of 100,000 cells/cm2 (at d0) and cultured for 24 hours at normal physiological culture conditions of 37°C and pH7.35 to facilitate cell attachment; subsequently, cells were cultured for an additional 48 hours at different temperatures or pH to investigate the effects of stresses on the adherent cell fraction and the cells in suspension at harvest (at d3). Numbers of cells were counted in a Neubauer Chamber, and are given as cells/cm2 and percent of controls (set as 100%), which were cells cultured at regular temperature of 37°C and regular pH of 7.35. Viabilities were determined by Trypan Blue exclusion, and they are given as percent viable cells. Data are given as means from n = 5 biological repeats ± standard deviation. Statistical significant differences of cells cultured under different nonphysiological conditions compared with control cultures under physiological conditions (cultures at 37°C or pH7.35, respectively) were determined by Student's t-test, with p ≤ 0.05; significantly changed expression is marked in bold.
Human fetal liver cells were precultured for 24 hours under normal culture conditions at 37°C and 5% CO2 to facilitate cell attachment, followed by a period of temperature or pH stress. We investigated the effects of stresses on adherent (parenchymal) as well as nonadherent (hematopoietic) cell types. In detail, we studied the effects on total cell number, viability, proliferation, gene and protein expression, as well as the effects on various cell type-specific fractions and their proliferation.
Cell numbers and viabilities
Temperature significantly influenced numbers of cells in culture (Table 1). Culture at a low temperature of 25°C after 3 days resulted in increased numbers of both adherent and nonadherent cells, to 122% and 206%, respectively, of controls at 37°C, whereas culture at 40°C decreased adherent cell numbers to 77% of controls but increased the nonadherent hematopoietic fraction to 165% of controls. Viabilities of neither adherent nor nonadherent cells were affected significantly by low- or high-temperature culture. When effects of low temperature were investigated on adherent cells in a longer-term culture of 7 days (Table 2), culture at 25°C resulted in a further increase in cell numbers, which was significantly higher for cells cultured at 25°C than at 37°C. Cell viabilities as determined by trypan blue exclusion assay were slightly higher in 25°C culture, but this difference was not statistically significant different; LDH activity, which can be measured in culture media if the enzyme is released by disintegrating cells, could not be detected in any of the cultures. We also compared proliferation rates at day 7 of these adherent cells at both culture temperatures, and we found that the percentage of Ki67+ cells in FACS was significantly higher at 25°C (15.1% ± 5.3%) than at 37°C (8.2% ± 1.9%) (p < 0.05).
Human fetal liver cells were initially seeded at densities of 100,000 cells/cm2 (at d0) and cultured for 24 hours at 37°C to ensure cell attachment. Subsequently, cells were cultured for additional 2 and 6 days at 25°C to investigate the effects of low-temperature stress on all adherent cells in longer-term culture (d7). Numbers of total cells were counted in a Neubauer Chamber, and are given as cells/cm2 and percent of controls (set as 100%), which were cells cultured at 37°C. Viabilities were determined by Trypan Blue exclusion given as percent viable cells, as well as LDH assay given as IU/L. Data are given as means from n = 4 biological repeats ± standard deviation. Statistical significant differences of cells cultured at 25°C compared with control cultures at 37°C were determined by Student's t-test, with p ≤ 0.05; significantly changed expression is marked in bold.
LDH, lactate dehydrogenase; n.a, not applicable.
Stress exerted by increased pH did not affect cell numbers or viabilities significantly when compared with controls (Table 1). Culture at low pH, however, resulted in significantly decreased numbers of adherent cells to 41% of controls but did not affect nonadherent cells significantly. None of the cell fractions were affected by low pH stress in their viabilities.
Flow cytometry for analyses of surface marker expressions and proliferation
We analyzed the effects of stresses on percentages and proliferation of specific liver cell populations in cultures of total fetal liver cells by fluorescence-activated cell sorting (Fig. 1). Cell surface markers characteristic of the various stem/progenitor types in the fetal liver also were used in combination with the nuclear proliferation marker Ki67. Surface markers included those that are characteristic for stem/progenitor cells of various lineages (SSEA4), hepatic stem/progenitor cells and bile duct epithelium (CD326), hepatoblasts (DLK1), mesenchymal and mesendodermal cells (CD90), nonerythrocyte hematopoietic cells (CD45), nonadherent erythrocytes and adherent erythroblasts (CD235a), hematopoietic stem cells (CD34), and endothelial cells (CD31, CD34). Because CD326 is expressed on hepatic stem cells as well as bile duct epithelium and no truly specific surface marker exists neither to distinguish between these nor to label uniquely hepatic stem cells, we additionally investigated the percentages of other hepatic stem/progenitor-cell-associated markers SSEA4, CD90, and DLK1 for their co-expression with CD326.
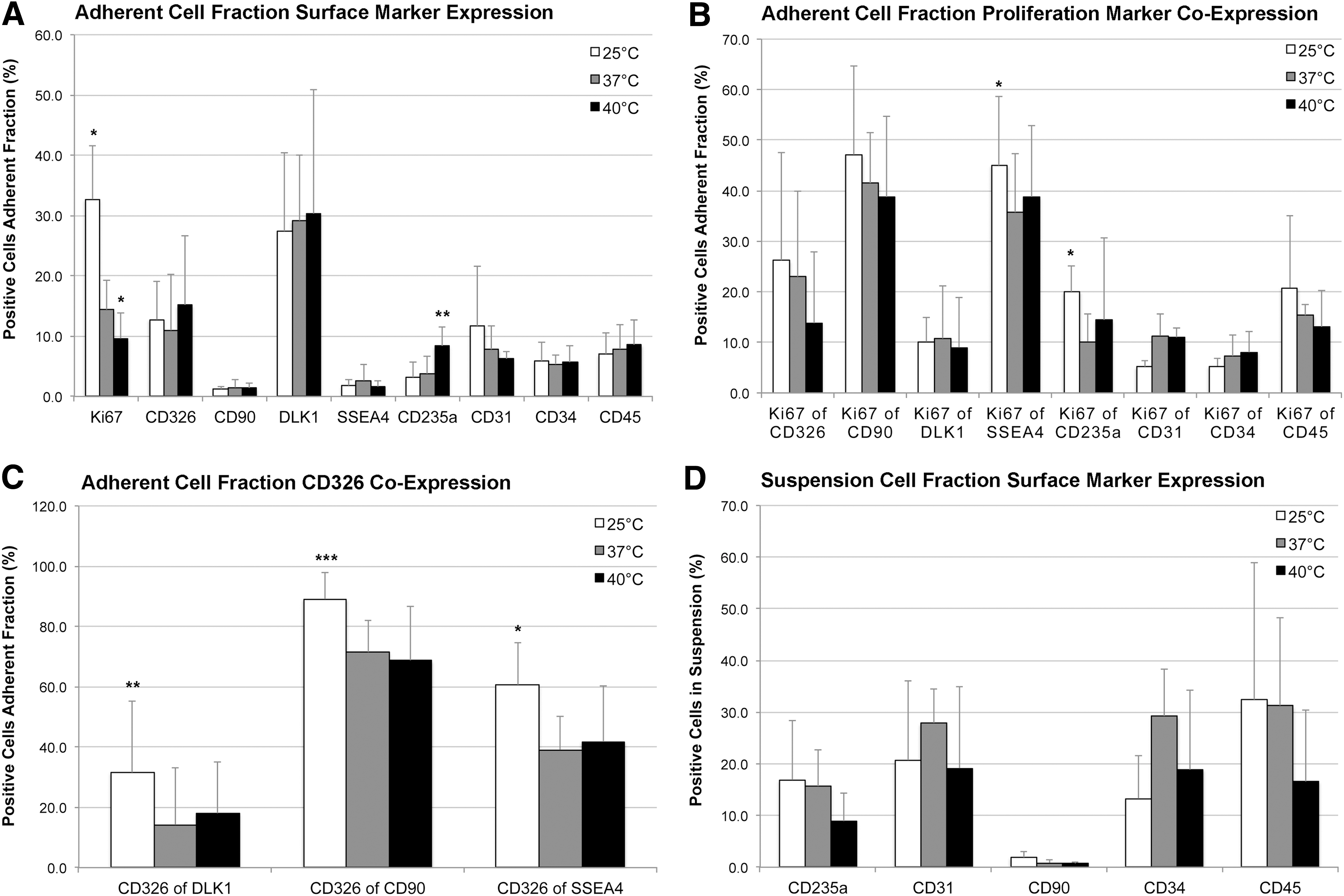
Effects of temperature stress on percentages and proliferation of human fetal liver cell populations after 3 days of culture. Human fetal liver cell fractions after culture at different temperatures of 25°C (white bars), 37°C (grey bars), or 40°C (black bars) were analyzed for proliferation and cell type-specific surface marker expression using FACS.
We found that low-temperature culture more than doubled the percentage of proliferating total adherent cells (Fig. 1), leading to a significant increase of 32.7% compared with controls at 14.4%. Higher proliferation rates (Ki67 positivity) of SSEA4+ stem cells and CD235a+ erythroblasts/erythrocytes could be observed. Also, significant increases could be detected in the percentages of cells co-expressing hepatic progenitor marker CD326 with SSEA4, CD90, or DLK1. High-temperature stress significantly reduced the percentage of proliferating adherent total cells, and increased the percentage of CD235a+ erythroblasts, but hepatic progenitor populations were not affected. Percentages of hematopoietic cells in suspension were not significantly affected by either low- or high-temperature stress.
Because we had observed significant effects of low temperature in cultures of total human fetal liver cells that resulted in increased cell numbers, proliferation, and stem cell-associated marker expression, we sought to investigate and distinguish specifically these effects on MACS presorted hepatic stem cells (selected for CD326+) and hepatoblasts (selected for DLK1+) (Fig. 2). Interestingly, we found that culture for 48 hours at 25°C significantly increased numbers (Fig. 2A) of CD326+ hepatic stem/progenitor cells but not DLK1+ hepatoblasts. Similarly, proliferation (Fig. 2B) of hepatic stem/progenitor cells (but not hepatoblasts) was also increased by low-temperature culture, but this difference was not statistically significant different.
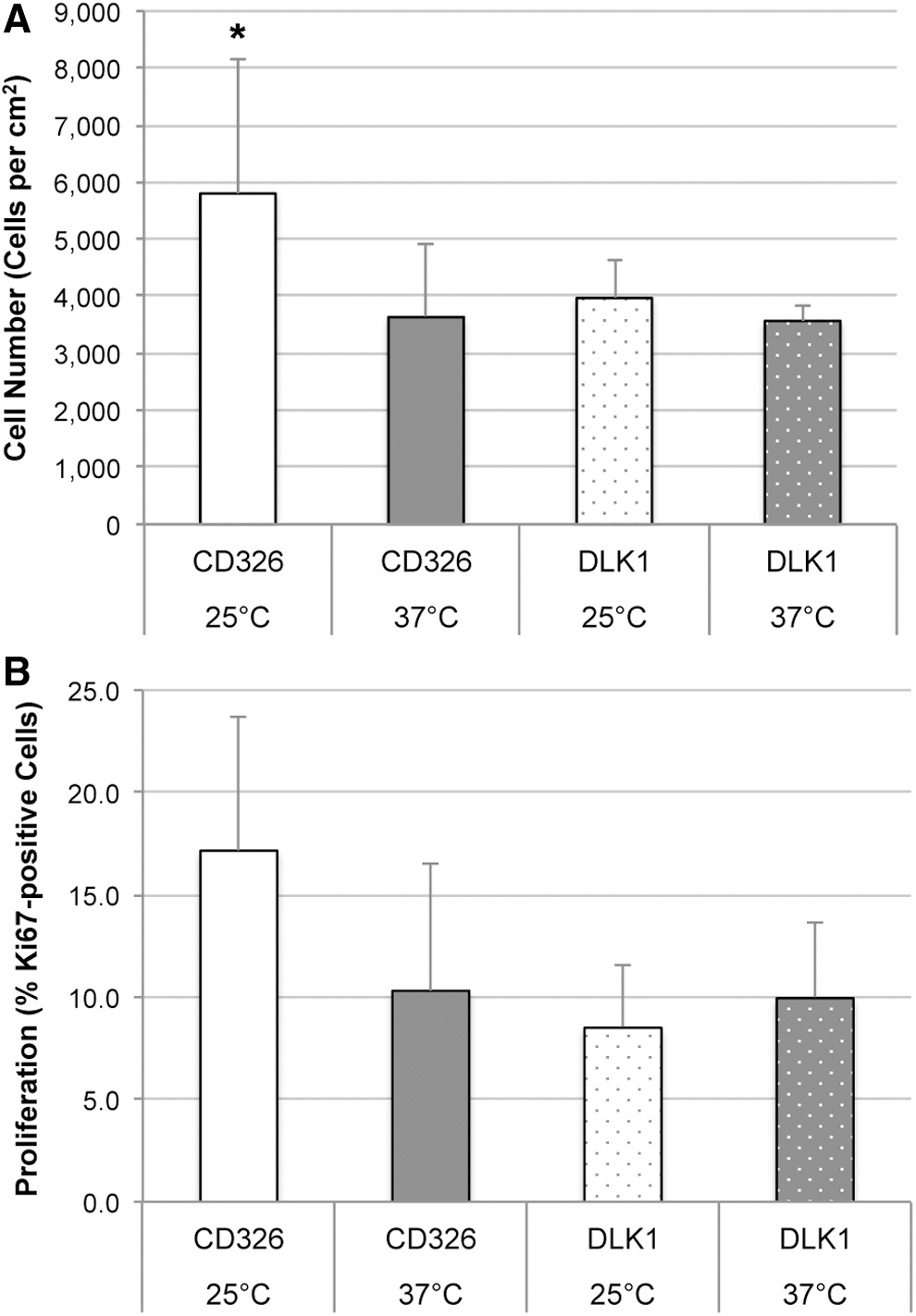
Effect of low-temperature stress on cell numbers and proliferation of presorted hepatic stem cells and hepatoblasts after 3 days of culture. Hepatic stem cells and hepatoblasts were sorted from freshly isolated total human fetal liver cell suspensions by using MACS for CD326+ (hepatic stem cells) and DLK1+ (hepatoblasts), respectively. Cells were seeded at 1000 cells per cm2, and after an initial attachment phase of 24 hours cells were subject to 48 hours of low-temperature stress at 25°C (white bars); controls were continuously cultured at 37°C (gray bars). At d3, cultures of hepatic stem cells (plain bars) and hepatoblasts (dotted bars) were analyzed for cell numbers
Stress triggered by high pH (Fig. 3) did not affect any of the adherent cells or cells in suspension. Low pH, however, caused significant effects on cell fractions. From the adherent cell fraction, the percentage of DLK1+ hepatoblasts increased, whereas the percentages of Ki67+ proliferating CD326+, SSEA4+, CD45+, and CD235a+ cells decreased. Hematopoietic CD45+ cells in suspension were significantly reduced.

Effects of pH stress on percentages and proliferation of human fetal liver cell populations after 3 days of culture. Human fetal liver cell fractions after culture at pH6.76 (white dotted bars), pH7.35 (gray dotted bars), or pH7.88 (black dotted bars) were analyzed for proliferation and cell type-specific surface marker expression by using fluorescence activated cell sorting.
Gene expression analyses
We investigated the effects of stresses on cell type-specific gene expression of adherent fractions in detail. Because the nonadherent fraction comprises mainly enucleated hematopoietic cells, we performed gene expression analyses only on adherent cells.
When cells were subject to temperature stress (Fig. 4), after 3 days in culture the expression of several genes changed significantly compared with controls kept at 37°C. Lower expression of mesenchymal (THY1, ACTA1, and ACTA2), mature hematopoietic (PTPRC), and mature endothelial (VWF) genes could be observed, and higher expression of serum protein (ALB and AFP), hepatoblast (DLK1), and multipotent (cancer) stem cell (POU5F1) marker genes; although these changes could be observed at both high and low temperature, these effects were more pronounced, especially under low-temperature culture. Some genes were expressed significantly different specifically in low- but not in high-temperature culture. These included genes associated with hepatic stem/progenitor cells (extremely lower expression of KRT19 and LGR5), endothelial and hematopoietic cells (higher expression of PECAM1 and CD34); also, the cell-cycle progression marker cyclin E1 was significantly upregulated in low-temperature culture but not under high-temperature culture. High-temperature culture resulted in reduced expression of some mesenchymal, endothelial, and hematopoietic genes but increased expression of hepatic genes albumin and DLK1. The effects of temperature on gene expression, in general, were also confirmed by significantly increased expression of known temperature-sensitive genes HSPA1A under both high and low temperature, as well as an expected, strongly increased expression of the CIRBP under low temperature and decreased expression under high temperature. When cells were kept for longer-term culture of 7 days (Fig. 5) at 25°C, the expression pattern of most genes was very similar to that of 3-day culture but even much more pronounced; note that data are given on logarithmic scale.
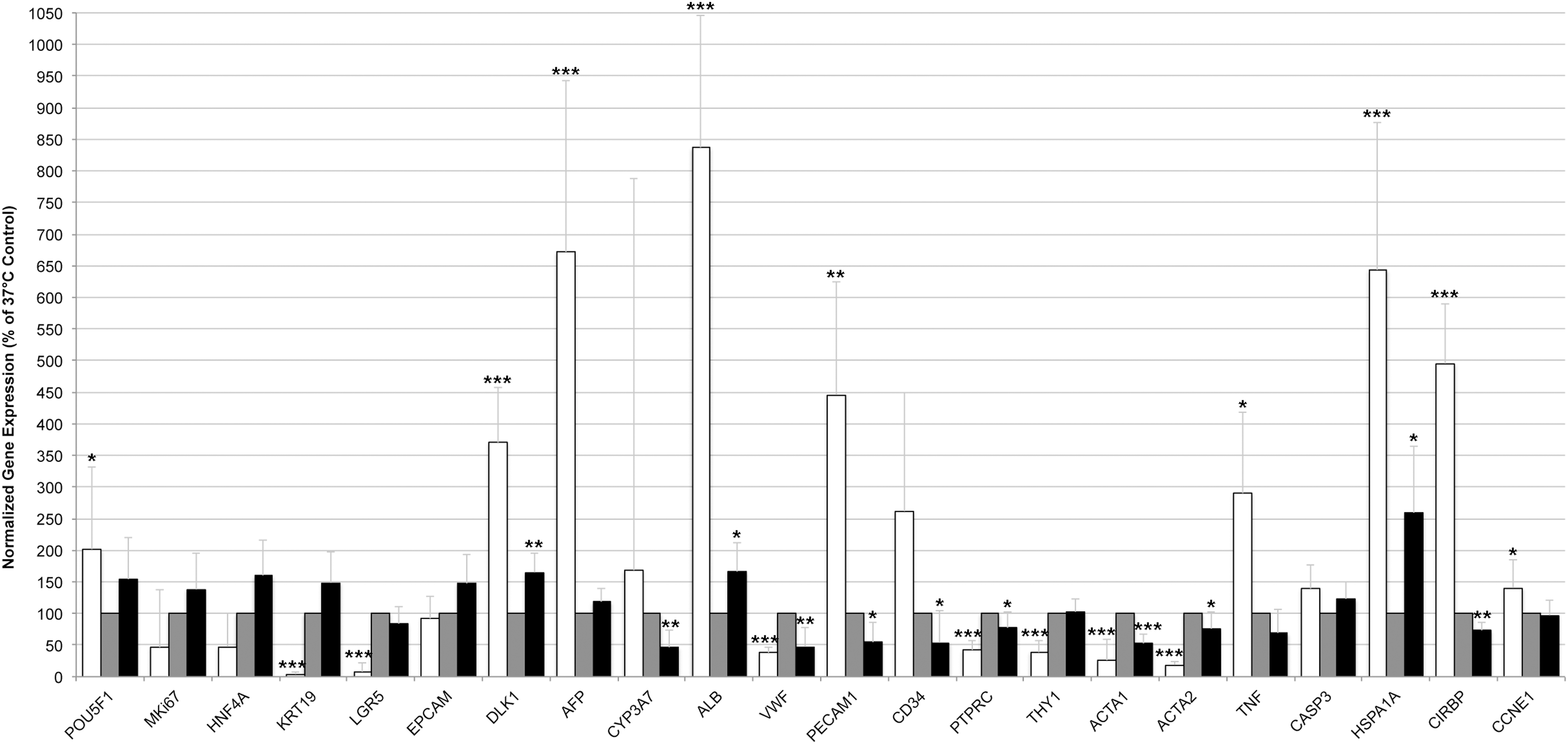
Effects of temperature stress on gene expression after 3 days of culture. Adherent human fetal liver cells after culture at different temperatures of 25°C (white bars), 37°C (gray bars), or 40°C (black bars) were analyzed for gene expression by real-time PCR. Gene expression was normalized to beta actin (ACTB), and cultures at regular physiological temperature of 37°C were used as reference (set as 100%). Data are given as means from n = 5 biological repeats ± standard deviation. Significant differences in gene expression of cells in culture at regular physiological temperature of 37°C versus high temperature of 40°C or low temperature of 25°C, respectively, were determined by Student's t-test with *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001. PCR, polymerase chain reaction.
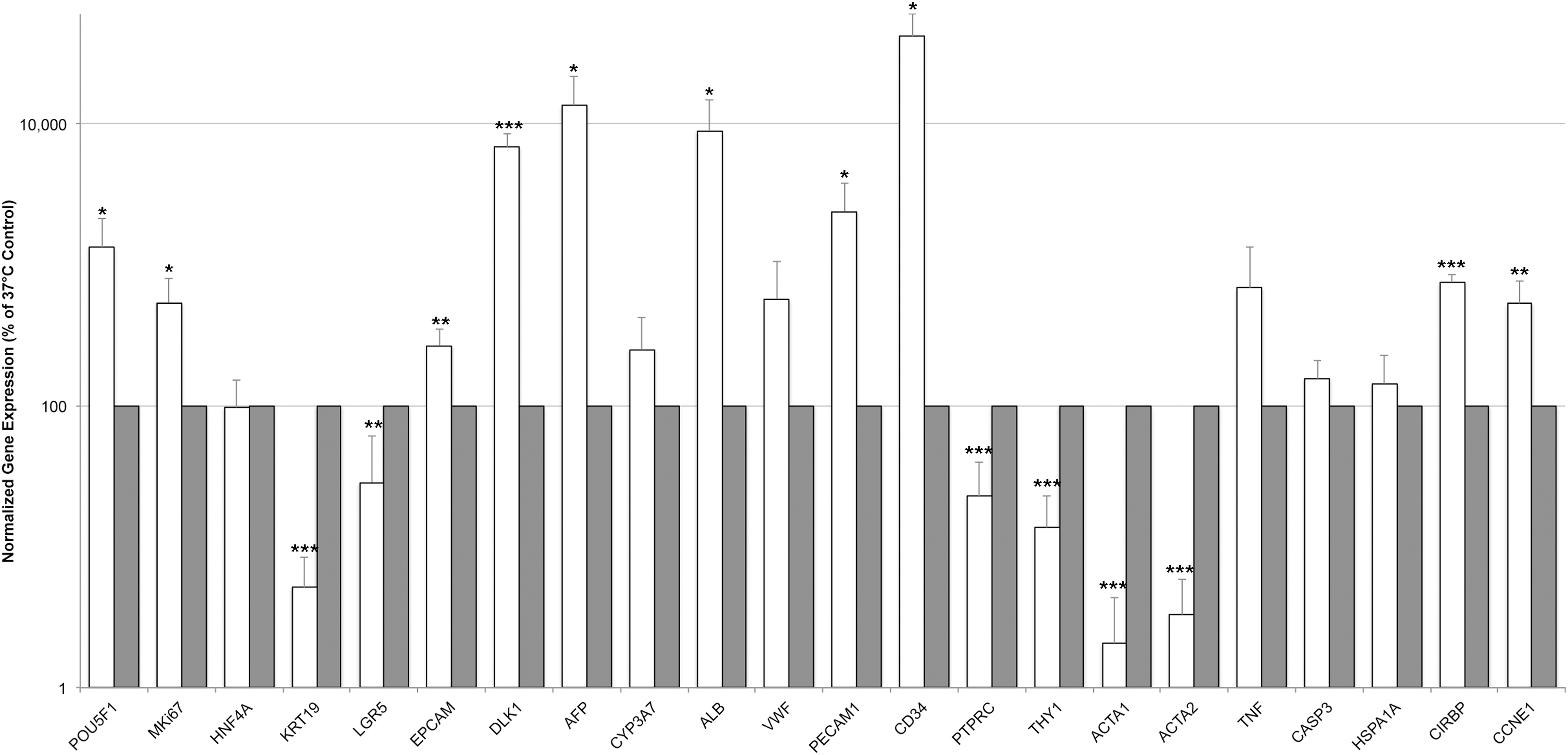
Effects of low-temperature stress on gene expression after 7 days of culture. After longer-term culture of 7 days, adherent human fetal liver cells in culture at 25°C (white bars) or 37°C (gray bars) were analyzed for gene expression by real-time PCR. Gene expression was normalized to beta actin (ACTB), and cultures at regular physiological temperature of 37°C were used as reference (set as 100%). Data are displayed on logarithmic scale, and given as means from n = 4 biological repeats ± standard deviation. Significant differences in gene expression of cells in culture at regular physiological temperature of 37°C versus low temperature of 25°C, respectively, were determined by Student's t-test with *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001.
Stress exerted by pH alteration affected gene expression moderately (Fig. 6). Surprisingly, culture at high pH did not substantially affect the expression of nearly all genes investigated. High pH significantly lowered expression of THY1, the gene encoding the mesenchymal surface marker CD90. In contrast, low pH significantly affected multiple genes in their expression. Interestingly, the pattern of expression changes was somewhat similar to that of cultures stressed by low temperature, for example, a strongly decreased expression of stem cell-associated genes KRT19 and LGR5, as well as that of ACTA1. Different from low-temperature stress, though, the expression of hematopoietic and mature endothelial (vWF) cell markers was not affected. Also, low pH did not influence the expression of TNF, whereas low-temperature stress increased the expression of TNF.
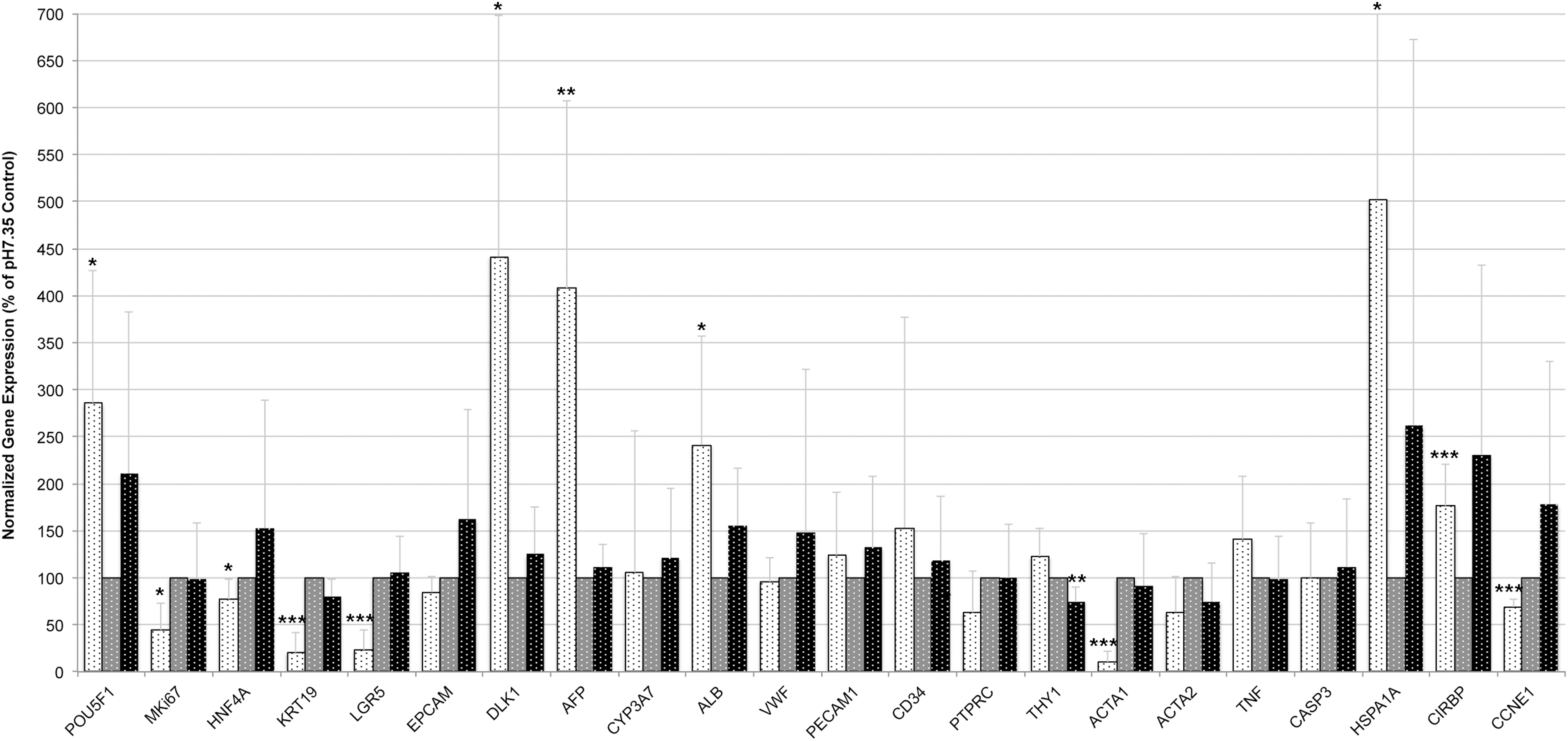
Effects of pH stress on gene expression after 3 days of culture. Adherent human fetal liver cells after culture at pH6.76 (white dotted bars), pH7.35 (gray dotted bars), or pH7.88 (black dotted bars) were analyzed for gene expression by real-time PCR. Gene expression was normalized to beta actin (ACTB), and cultures at regular physiological pH were used as reference (set as 100%). Data are given as means from n = 5 biological repeats ± standard deviation. Significant differences in gene expression of cells in culture at regular physiological pH7.35 versus high pH7.88 or low pH6.76, respectively, were determined by Student's t-test with *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001.
Immunocytochemistry
We had observed significant increases in cell number, proliferation, and stem cell-associated marker expression by flow cytometry, but higher expression of mature hepatic associated genes (ALB, AFP) and lower expression of hepatic stem cell associated genes (KRT19, LGR5) in low-temperature culture. Gene expression analysis is, however, reflective of the total cell population, and it does not give information on the different populations of the liver. Also, gene expression does not necessarily correlate with protein expression, which is especially true for secreted proteins such as albumin and alpha-fetoprotein that provide a feedback loop on gene expression. Therefore, we further sought to examine adherent cells that had been exposed to temperature stress for their protein expression by using immunocytochemistry (Fig. 7). We investigated the co-expression of alpha-fetoprotein and cytokeratin 19, because cytokeratin 19 is expressed in hepatic stem/progenitor cells and bile duct epithelium but diminishes in hepatoblasts, the progeny of the hepatic stem/progenitor cells; whereas alpha-fetoprotein is strongly expressed in hepatoblasts but not in hepatic stem/progenitor cells or bile duct epithelium. Thus, co-staining allows distinguishing between these two cell lineages.
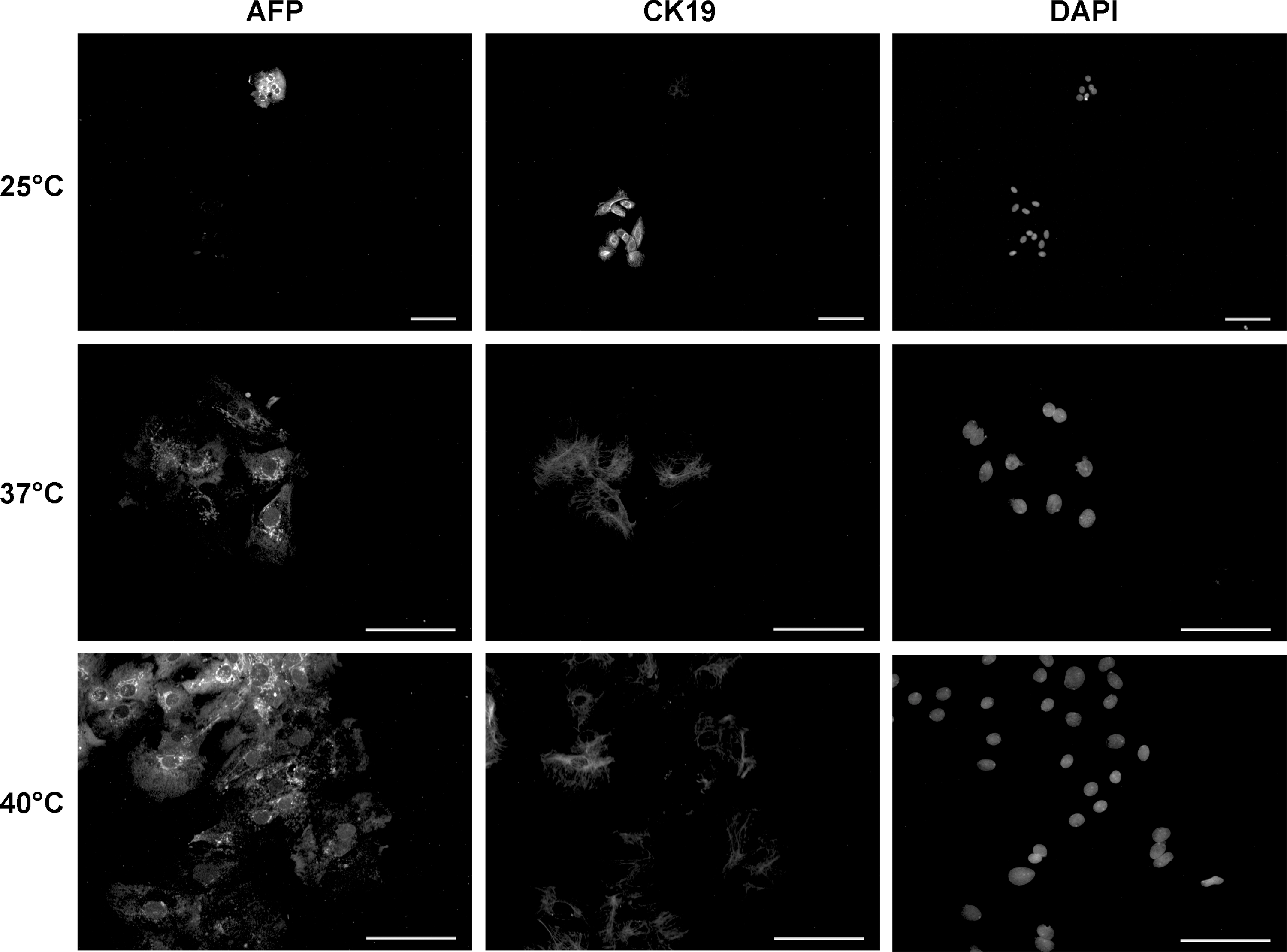
Immunocytochemistry of human fetal liver cells exposed to temperature stress after 3 days of culture. Cell cultures were kept at temperatures of 25°C, 37°C, or 40°C for 48 hours, and fixed cells were stained for alpha-fetoprotein (AFP) and cytokeratin 19 (CK19) proteins. Cell nuclei were stained with DAPI. Photographs of cultures at 25°C are given at 100 × magnification to display the two different types of colonies; photographs of cultures at 37C° and 40°C are given at 200 × magnification to show details of the prevalent colony type. All scale bars: 100 μm. DAPI, 4′,6-diamidino-2-phenylindole.
Cultures at 37°C or 40°C revealed morphologically typical hepatic stem/progenitor cell colonies, with most cells noticeably expressing alpha-fetoprotein, and most, but not all, cells also expressing moderate levels of cytokeratin 19. At 40°C, cell morphology appeared more flattened, with cells having an increased diameter. Interestingly, in culture at 25°C, we observed two types of colonies: either colonies formed out of cells strongly positive for CK19 but negative for hepatoblast marker AFP (54 ± 11%), or colonies containing cells strongly positive for AFP but negative for CK19 (35% ± 6%), but very few cells co-expressing AFP and CK19 (11% ± 4%), indicating that low-temperature culture promotes early hepatic stem cell maintenance and proliferation.
Flow cytometry for relative analyses of cell size
Because the size of cells in culture seemed to be affected by temperature as observed in immunocytochemistry, we compared their relative size distribution in flow cytometry by using forward scatter histograms. Examples of single histograms with gating are given in Supplementary Figure S1. In general, we observed a peak of smaller cells followed by a wider distribution of larger cells, where a comparative gate was set. Smaller cell types comprise mostly hematopoietic cells and stem/progenitor cells; this is also reflected by the fact that the cells in suspension comprised a larger fraction of smaller cells than the adherent cells. By average (n = 3 biological repeats), adherent cells in culture at 25°C and 40°C comprised an increased fraction of cells with larger sizes (65.7% ± 3.9% and 68.7% ± 1.4%, respectively) compared with controls at 37°C (64.0% ± 10.9%), but these differences were not statistically significant. Cells in suspension in culture at 25°C comprised a decreased fraction of cells with larger sizes (34.2% ± 2.7%) compared with controls at 37°C (42.5% ± 8.7%), and cultures at 40°C had an increased fraction of cells with larger sizes (48.2% ± 5.8%); however, these differences were not statistically significant different.
Discussion
Longstanding data have demonstrated a direct correlation of culture temperature with cell proliferation; lower temperature has been traditionally associated with less proliferation. For example, mouse leukemic cells slowed down their growth rates with decreasing temperatures from 40°C to 28°C 3 ; a human amnion cell line demonstrated similar highest DNA synthesis rates between 37°C and 39°C but strongly decreased rates below and above these optima 17 ; and a rat hepatoma cell line responded to 25°C culture with complete cell-cycle arrest and stop of proliferation. 18 In general, metabolic reactions slow down below 37°C, because the optimum temperature for the kinetics of most enzymes in mammals is about 40°C. Similarly, the kinetics of many mammalian enzymes are decreased below or above the physiological pH optimum of about 7.4. Mammalian cells respond to thermal stress below or above the physiological optimum (of usually 37°C) with induced gene expression of various HSPs, which, in turn, activate different pathways involved in apoptosis, growth arrest, and differentiation. 4 –6 Culture at temperatures lower than 37°C has been shown to arrest cell-cycle progression in G1, G2, or M phase, depending on the actual cell type and temperature, 17 –19 and to induce CIRBPs. 20
Interestingly, in our research, we found that fetal hepatic stem/progenitor cells respond to low-temperature culture with increased proliferation and cell numbers, which is in contrast to those data that have been published to date on adult normal and tumorigenic cell types. Effects of low-temperature stress on human liver stem/progenitor cells have not been investigated, but hepatic stem/progenitor cells are resistant to cold ischemia, 7,8 as viable and prolific stem/progenitor cells could be isolated and cultured in the long term successfully from adult intact livers that had been subject to 48 hours 7 or 24 hours to even 168 hours 8 ischemia at 4°C. Of interest is also that temperature seems to have an influence on the balance of mesenchymal versus hepatic fate. At a decreased temperature of 25°C, typical hepatic genes such as albumin, AFP, DLK1, and CYP3A7 were significantly increased; whereas mesenchymal genes such as THY1, ACTA1, and ACTA2 were strongly reduced. This observation relates to findings from Shui and Scutt, where an increased temperature of 39°C induced proliferation and mineralization of human bone marrow mesenchymal stromal cells. 10 Temperature could have an influence in epithelial-mesenchymal transition, 21 playing a role in liver fibrosis, 22 or recruitment of extra-hepatic mesenchymal stem cells. 23
Activation of proliferation of hepatic stem/progenitor cells by moderately low temperatures could be potentially initiated through induction of CIRBPs. CIRBP activation by a moderately low temperature of 32°C has been shown to conserve stemness and to suppress apoptosis of neural stem cells. 24 Recent findings by Sakurai et al. using knockout mice demonstrated that the expression of CIRBP is positively correlated with the development and recurrence of hepatocellular carcinomas. 25 It remains to be investigated in detail as to whether CIRBP is a major activator of proliferation by low temperature in hepatic stem/progenitor cells; indeed, based on our data, we could observe significantly increased CIRBP gene expression at 25°C culture. In most mammals, certain cell types are naturally exposed to a considerably lower temperature than that of the body core, in particular, those of skin and testes. Notably, both contain stem/progenitor cell types with high proliferative capacity, with male spermatogenesis being highly sensitive to increased temperature, even that of 37°C (for a review, see Mieusset and Bujan 26 ). Taken together, these and our data indicate that stem/progenitor cell types respond to moderately low temperature much differently than mature cell types, being induced to proliferate instead of growth arrested.
In addition to temperature stress, we also investigated the effects of un-physiological low and high pH on liver stem/progenitor cells. Forty-eight hours exposure to high pH did not cause major effects on cell numbers, proliferation, or gene expression of any liver cell type, which is different from cultures of human bone marrow mesenchymal stem cells that have been shown to have the highest proliferation rates at highly alkaline pH8.0. 11 Osteoclasts, on the other hand, generate an acidic environment by secretion of protons, resulting in bone resorption, 27 so the optimum pH appears to be cell type specific. Exposure of human fetal liver cells to low pH resulted in significantly decreased cell numbers and proliferation of the adherent, hepatic stem/progenitor cell fraction as well as hematopoietic mature CD45+ cells and CD235a+ adherent erythroblasts. Gene expression analyses demonstrated reduced expression of typical hepatic progenitor genes and increased expression of mature hepatic genes. These results indicate that low pH stimulates hepatic maturation rather than hepatic proliferation, and it simultaneously suppresses hematopoiesis. However, POU5F1, which is a typical gene expressed by early multipotent embryonic stem cells as well as cancer stem cells and codes for Oct-4 protein, was increased in acidic culture.
In vivo, an acidic microenvironment has been detected in many tumors (for a review, see Kato et al. 28 ), in which lactate secretion due to anaerobic glycolysis lowers the pH. The hypoxic environment in tumors (resulting in acidic pH) can increase the expression of Oct-4, which, in turn, leads to de-differentiation and the formation of cancer stem cells, as has been demonstrated in melanoma. 29 In nonpathological conditions, a low pH can be observed in the extracellular environment of several tissues and is naturally well tolerated by adapted cells, for example, those of the epidermal layers of skin, 30 vagina, 31 and lung (for a review, see Fischer and Widdicombe 32 ), thereby also protecting against bacterial infections. In adult liver, the secretion of bile acids could contribute to local acidification; although the majority of bile acids are salts because they exist as conjugates, about 10% of bile acids in humans are produced via an alternative pathway, giving rise to acidic intermediates (for a review, see Li and Apte 33 ). Certain bile acids have been implicated in liver regeneration by supporting regeneration through receptor activation (for a review, see Fan et al. 34 ), but the implication of bile acids' acidity is unclear.
Modifying cell culture temperature could provide a simple and cost-effective means for the enhanced expansion of liver cell populations. In vitro expansion of cell numbers is of great interest for therapeutic applications in clinical cell-based liver therapies, such as extracorporeal liver support 35,36 or liver cell transplantation. 37 –41 Although we focused on stem/progenitor liver cell cultures in this study, it could be of interest to investigate similar responses in other cell types as well.
In conclusion, we found distinct effects of acidic pH and low temperature on specific stem/progenitor cell types of the human fetal liver. Low pH stimulated hepatic maturation and suppressed hematopoiesis. Interestingly, culture at a low temperature of 25°C increased cell numbers and proliferation of adherent, hepatic stem/progenitor cells—which is in contrast to data known to date of adult normal and tumorigenic cell types—indicating that moderate low-temperature stress specifically induces stem/progenitor cell proliferation. These findings could also have implications for the expansion of stem/progenitor cell populations for clinical applications.
Footnotes
Acknowledgments
This study was financially supported by the University of Pittsburgh Medical Center. The authors thank Lynda Guzik of the FACS Core at the McGowan Institute and the staff of the Allegheny Reproductive Health Center for their continuous support.
Authors' Contributions
E.S.: Conception and design, collection and assembly of data, data analysis and interpretation, article writing, and final approval of the article; H.G.F. and R.L.T.: Provision of study material, final approval of the article; A.L.: Administrative support, final approval of the article; B.G. and J.C.G.: Financial support, final approval of the article.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.