
Abstract
Mitochondrial structural and functional integrity is maintained through the coordination of several processes (e.g., biogenesis, dynamics, mitophagy), collectively referred to as mitochondrial quality control (MQC). Dysfunctional MQC and inflammation are hallmarks of aging and are involved in the pathogenesis of muscle wasting disorders, including sarcopenia and cachexia. One of the consequences of failing MQC is the release of mitochondria-derived damage-associated molecular patterns (DAMPs). By virtue of their bacterial ancestry, these molecules can trigger an inflammatory response by interacting with receptors similar to those involved in pathogen-associated responses. Mitochondria-derived DAMPs, especially cell-free mitochondrial DNA, have recently been associated with conditions characterized by chronic inflammation, such as aging and degenerative diseases. Yet, their actual implication in the aging process and muscle wasting disorders is at an early stage of investigation. Here, we review the contribution of mitochondria-derived DAMPs to age-related systemic inflammation. We also provide arguments in support of the exploitation of such signaling pathways for the management of muscle wasting conditions.
Introduction
M
Due to their relevance to cell physiology and whole-body metabolism, a comprehensive set of adaptive quality control mechanisms is in place to ensure the preservation of mitochondrial structural and functional integrity. 4 Mitochondrial quality control (MQC) mechanisms also allow for the dynamic modulation of organelle function and number to meet the heterogeneous energy demands of the various tissues. MQC is accomplished through a set of interrelated processes (i.e., protein folding and degradation, mitochondrial autophagy, mitochondrial fission and fusion, and mitochondrial biogenesis). 4,5
The regulation of mitochondrial content is achieved through the dynamic balance between mitochondrial biogenesis and degradation. Mitochondrial biogenesis is a multistage process finalized to producing new mitochondria upon the coordinated expression of nuclear and mtDNA-encoded genes. The process is primarily orchestrated by members of the peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family, namely PGC-1α and PGC-1β (reviewed in Picca and Lezza 6 ). On the other hand, mitochondrial dynamics, through the coordination of fusion and fission, control organelle shaping, metabolic plasticity, redox signaling, and cell death/survival pathways. 7 –9 Mitochondrial fusion allows for mtDNA mixing within the network, thereby preventing focal accumulation of mutant mtDNA and preserving mtDNA integrity. 10,11 Mitochondrial fission, instead, segregates defective or unnecessary organelles for their subsequent removal through mitophagy. 4 The integration of mitochondrial dynamics with the selective removal of dysfunctional mitochondria, referred to as mitophagy, ensures an efficient MQC process and preserves metabolic cellular “fitness.” 12
Derangements of the MQC axis have been described during aging and in the context of a number of disease conditions, including cancer, cardiovascular disease, diabetes, and neurodegenerative disorders. 13,14 Along with mitochondrial dysfunction, chronic inflammation is a hallmark of both aging and degenerative diseases. The two phenomena may be linked to one another. Indeed, emerging evidence indicates that circulating cell-free mtDNA, one of the damage-associated molecular patterns (DAMPs), may establish a functional relationship between mitochondrial damage and systemic inflammation. 15 –18 mtDNA can be released into the circulation in response to cell insults. Here, it is able to induce an inflammatory response through hypomethylated CpG motifs resembling those of bacterial DNA. 19 These regions, indeed, bind and activate membrane or cytoplasmic pattern recognition receptors (PRRs), such as the Toll-like receptor (TLR), the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR), 20 and cytosolic cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) DNA sensing system-mediated pathways. 21
The role of dysfunctional MQC in systemic inflammation is exemplified by the involvement of neutrophils in the pathogenesis of systemic lupus erythematosus (SLE). 16 Neutrophils are constitutively unable to remove damaged mitochondria via mitophagy. However, through a complex response triggered by specific autoantibodies, neutrophils of SLE individuals compensate such a defect by two complementary pathways: (1) extrusion of mitochondrial components (e.g., mitochondrial transcription factor A [TFAM] and mtDNA), and (2) exportation of oxidized mtDNA, upon dissociation from TFAM, into lysosomes for degradation. 14 Here, oxidized mtDNA activates the TLR pathway.
The possible contribution of mitochondrial DAMPs to the inflammatory milieu that characterizes muscle wasting disorders has not yet been explored. However, this hypothesis is worth being pursued as it could help identify novel biological targets for the management of muscle loss.
Here, we summarize the current evidence on circulating mtDNA as a trigger for age-related systemic inflammation. We first describe two candidate mechanisms generating and releasing cell-free mtDNA: (1) dysregulation of TFAM binding to mtDNA, and (2) impairment of mitophagy. Subsequently, we illustrate the pathways linking mitochondrial dysfunction with systemic inflammation during aging. Finally, we propose a role for the triad “MQC failure/cell-free mtDNA/inflammation” in two major muscle wasting disorders (sarcopenia and cachexia).
The Interaction of TFAM with mtDNA as a Modulator of Mitochondrial Biogenesis
Mitochondrial biogenesis is a complex, multistage process that converges on the activation of several transcriptional coactivators and culminates in the generation of newly synthetized organelles. Several (patho)physiological conditions, such as exercise, fasting, oxidative stress, and inflammatory cell stress, can induce mitochondrial biogenesis that, depending on the stimulus, is achieved through the activation of distinct signaling pathways. 22 Relevant transcriptional coactivators are those belonging to the PGC-1 family (PGC-1α and PGC-1β), the nuclear respiratory factor (NRF) 1 and 2, and the estrogen-related receptor alpha (ERRα), which regulate the expression of mitochondrial proteins encoded by nuclear DNA (reviewed in Picca and Lezza 6 ). Mitochondrial biogenesis is regulated by transcription factors and coactivators through feedback loops involving, for instance, reactive oxygen species (ROS), calcium, and anti-inflammatory signaling (reviewed in Dominy and Puigserver 23 ), and the fine modulation of gene expression at the level of regulatory elements. Subsequently, an increase in the expression of several mitochondrial proteins, including those binding the mtDNA (e.g., TFAM and mitochondrial transcription factors B1 and B2 [TFB1M and TFB2M]), occurs. 24,25 These mediators, like the majority of mitochondrial proteins, are encoded by the nuclear genome and transported into mitochondria via a protein import system. 26,27 Once entered the mitochondrion, mtDNA-binding proteins directly activate mtDNA transcription and replication through their interaction with mtDNA.
Mammalian mtDNA, a circular double-stranded DNA ∼16.5 kb-long, encodes for 2 ribosomal RNAs, 22 transfer RNAs, and 13 protein subunits of the electron transport chain (ETC). The mtDNA also contains two noncoding regions (NCRs): (1) the major NCR, namely the D-loop, which encompasses the transcription promoter of both heavy and light strands (HSP1 and HSP2, LSP) and the origin of replication of the heavy strand (OriH), and (2) the minor NCR, which includes the origin of replication of the L strand (OriL). The D-loop region is the major site of transcriptional regulation, as reflected by its interaction with multiple regulatory proteins. 28
mtDNA is organized into nucleoids, protein-DNA complexes, residing within the mitochondrial matrix. 29 TFAM represents one of the prominent components of such complexes bound at the level of the inner mitochondrial membrane. 30 This transcription factor belongs to the high-mobility-group (HMG) proteins and is able to bind, unwind, and bend mtDNA without sequence specificity. However, a preferential interaction with some regions of the mtDNA has recently been reported. 31 Through its binding, TFAM modulates several processes, including mtDNA replication and transcription, mtDNA maintenance, and possibly mtDNA repair (reviewed in Picca and Lezza 6 ). Recently, studies employing in vivo binding analysis of TFAM to specific mtDNA regions have shown that mitochondrial biogenesis is also regulated through TFAM-mtDNA interactions. 6,32 Dysregulation of this interaction has been reported during aging as a result of changes in the expression, turnover, and binding of TFAM secondary to TFAM and/or mtDNA alterations. 32,33 The relevance of this mechanism to muscle wasting disorders is discussed further in the article.
Oxidative Stress and mtDNA Alterations
Mitochondria are the major source of ROS, the generation of which occurs at distinct organelle sites as a by-product of substrate oxidation and oxidative phosphorylation (reviewed in Brand 34 ). According to the “mitohormesis theory,” when present in moderate amount, ROS function as intracellular signaling molecules that may improve systemic defense mechanisms by inducing an adaptive response. 35 Particularly relevant is the hydrogen peroxide cell-warning system for oxidative stress, which operates through its leakage from mitochondria and acts as a retrograde signal to nuclear-targeted cytosolic pathways. 36 However, when intracellular ROS concentrations overwhelm antioxidant defenses, cell homeostasis becomes compromised.
Mitochondrial constituents are primary targets of oxidative stress. 37 In particular, mtDNA, although packaged into nucleoids, is located in close proximity to the ROS source (i.e., the ETC). As a consequence of increased ROS generation, mtDNA can undergo several qualitative and/or quantitative alterations, including bases modifications, abasic sites, single- and double-strand breaks, point mutations, and large-sized deletions. 37,38 Besides damaging mitochondrial macromolecules, oxidative stress can also result in loss of ROS signal localization, 39 further contributing to the disruption of cell homeostasis.
Particularly interesting are the differential outcomes of redox imbalance in inflammation. In the setting of moderate inflammation, if cellular repair systems are overwhelmed the intrinsic apoptotic cascade may be triggered. 40 In the context of severe inflammation, mitochondrial dysfunction and ROS-induced damage may instead drive necrosis, leading to the release of cellular contents, including whole and fragmented mitochondria. As discussed in the next section, these events represent powerful inflammatory stimuli.
Mitochondrial Dysfunction and Inflammation
Although considered for long time a defense response against microbial agents, inflammation can also occur in the absence of infections. This is the case of the (chronic) inflammatory response referred to as “sterile inflammation.” 41,42
Local and systemic inflammation responses are mounted through the recruitment and activation of distinct macrophage subsets. Indeed, while tissue-resident macrophages serve anti-inflammatory functions to safeguard tissue homeostasis and resolve local inflammation, circulating monocytes are proinflammatory subtypes that limit the spread of the infection. 19 Similarly, sterile inflammation is organized within the framework of the innate immune response but is triggered by misplaced cellular components, including those of mitochondrial origin, that are subsequently disposed of by phagocytes.
Mitochondria contribute substantially to the sterile inflammatory process through the activation of several pathways. 43,44 Among these, the TLR pathway is regulated through redox-sensitive inflammatory signaling based on mitochondrial calcium handling, ROS production, and nuclear factor κB (NF-κB) activation. In conditions of calcium overload, such as burn injury or sepsis, 45 the ETC becomes dysfunctional and excessive ROS generation occurs. Such a ROS burst represents a major proinflammatory stimulus through the modulation of the expression and activity of NF-κB. 43
In addition to this, Matzinger 46 proposed mitochondria as contributors to sterile inflammation through a pathway deeply rooted into the “danger theory” of inflammation. According to this theory, the accumulation of DAMPs released from injured cells are able to induce caspase-1 activation and the release of proinflammatory cytokines. 47 In this context, cell-free mtDNA, N-formyl peptides, and cardiolipin are mitochondrial-derived DAMPs released in response to cell damage and death, able to activate inflammation. 46,48 Noticeably, degraded mtDNA has recently been identified as a DAMP subtype and a possible trigger of neurodegeneration. 49 Due to their bacterial ancestry, mitochondrial DAMPs can bind and activate membrane or cytoplasmic PRRs similar to those recognized by pathogen-associated molecular pattern (PAMPs). 47 The TLR pathway is eventually triggered by the binding of DAMPs to neutrophils, which, in turn, become activated and modulate the inflammatory response via NF-κB signaling 43 (Fig. 1).

Damaged mtDNA can trigger inflammation through three distinct signaling pathways by interacting with (1) TLRs, (2) NLRP3 inflammasome, and (3) cytosolic cGAS-STING DNA sensing system. cGAS, cyclic GMP-AMP synthase; IFN, interferon; IL, interleukin; IRF-1, interferon regulatory factor 1; mtDNA, mitochondrial DNA; NF-κB, nuclear factor κB; NLRP3, NLR family pyrin domain containing 3; NOD, nucleotide-binding oligomerization domain; ROS, reactive oxygen species; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; TLRs, Toll-like receptors; TNF-α, tumor necrosis factor alpha.
mtDNA is also able to induce an alternative pathway operating through the NLR family pyrin domain containing 3 (NLRP3) inflammasome. 50 –54 Given its association with several inflammatory diseases, NLRP3 is one of the most studied NLRs. NLRP3 consists of a group of cytosolic protein complexes, the activation of which results in the immediate engagement of caspase-1. The latter subsequently cleaves and activates the inactive precursors of interleukin (IL) 1β and 18 55 (Fig. 1). Interestingly, the redox-sensitive inflammatory and inflammasome-mediated pathways may act synergistically to reinforce the inflammatory response. 56
An additional component of the innate immune system is the cGAS-STING DNA sensing pathway. cGAS, upon binding to mtDNA, proceeds with STING protein recruitment that triggers the phosphorylation of the transcription factor interferon regulatory factor 3 (IRF-3) via TANK-binding kinase (TBK). Activated IRF-3 induces the production of type I and type III interferons (IFNs; β and λ1) and IFN-stimulated nuclear gene products (Fig. 1).
Circulating Mitochondrial DAMPs: Much More Than Waste-Derived Molecules
In agreement with the original “danger theory” of inflammation, 46 several syndromes characterized by systemic inflammatory response, including trauma, HIV and cancer, have been found to be associated with increasing levels of circulating mitochondrial DAMPs. 15,57,58
Interestingly, together with proinflammatory cytokines (e.g., IL6, tumor necrosis factor alpha [TNF-α], Regulated on Activation Normal T Cell Expressed and Secreted [RANTES], and IL1 receptor antagonist), circulating levels of mtDNA molecules have been found to increase past the age of 50. 59 In this context, the finding of increased TNF-α production following exposure of monocytes to mtDNA concentrations similar to those detected in vivo supports the idea of circulating mtDNA as factor contributing to systemic inflammation during aging (“inflamm-aging”). 59
TFAM has recently been proposed to act as a mitochondrial DAMP both in rodents and humans. 60 Mouse embryonic fibroblasts expressing only one TFAM allele show a 50% decrease in mtDNA content associated with constitutive activation of the cGAS-STING-IRF3 pathway. 61 Moreover, TFAM acts as a specific DAMP-inducing proinflammatory and cytotoxic response in in vitro models of human brain microglia. 60 In addition, TFAM appears to be relevant during inflammation through the modulation of its binding to mtDNA. Indeed, TFAM has been involved in rerouting oxidized mtDNA to lysosomes for degradation in neutrophils. 16 The extrusion of oxidized nucleoids by neutrophils in SLE represents a powerful immune system activator. 16
Systemic inflammation is a recognized feature of several musculoskeletal disorders. Recently, a fracture-initiated systemic inflammatory response syndrome (SIRS), characterized by increased circulating levels of cell-free mtDNA, has been documented in patients with hip fracture. 62 In such a context, circulating mtDNA appears to promote the development of inflammation by recruiting leukocytes. 62 It is noteworthy that specific alterations of the MQC axis have been identified in muscle biopsies obtained from old hip-fractured patients. 63 Taken together, these findings suggest the existence of a functional link between muscular mitochondrial dysfunction and systemic inflammation, possibly mediated by the release of mtDNA into the circulation. Conversely, moderate aerobic exercise, a well-known anti-inflammatory intervention, 64 decreases systemic cell-free mtDNA levels in healthy adults. 65
Despite the evidence supporting cell-free mtDNA and TFAM-bound mtDNA as DAMPs, the exact mechanism of mtDNA delivery into the cytosol and then into the circulation is currently unknown. As elegantly reviewed by Safdar et al., 66 one of the mechanisms through which eukaryotic cells communicate with each other is a signaling system relying on the exocytosis of proteins containing secretion-targeting sequences. 67 Being too labile within the extracellular environment, proteins and other macromolecules (including mtDNA), may be secreted within small membranous extracellular vesicles. 68 –70 A system of vesicles called exosomes is thought to use such a pathway to release a set of molecules (exerkines) in muscle under endurance exercise. 66 Exerkines contribute to mediating the beneficial effect of exercise by allowing systemic adaptations through autocrine, paracrine, and/or endocrine signaling. 66 Interestingly, cell-free mtDNA has been identified among the molecules released via exosomes. 66 Regardless of the actual mechanisms generating and releasing DAMPs, which goes beyond the purpose of this review, their accumulation has been shown to activate tissue resident macrophages and also favor tissue leukocyte infiltration. 71
The involvement of mitochondrial dysfunction and inflammation in the pathogenesis of muscle wasting disorders is well established. However, if and how mitochondrial DAMPs are involved in the inflammatory response associated with those conditions is presently unknown. The existence of crosstalk between mitochondria and the inflammasome is a highly promising area of investigation, especially in the context of muscle wasting disorders.
Dysfunctional Mitochondrial Quality Control and DAMPs Release
Autophagy is a cellular self-eating process whereby intracellular components are degraded within lysosomes during periods of stress (e.g., nutrient deprivation) as an attempt to adapt and survive. 72 Mitophagy, instead, involves the selective autophagic removal of mitochondria and is triggered by the loss of mitochondrial membrane potential. 73 This selective elimination, by clearing dysfunctional organelles, limits ROS generation and preserves cell viability. 74 However, mitophagy, is an extreme attempt of the cell to maintain homeostasis since mitochondria can dispose damaged components through an alternative route. In fact, before wholesale organelle degradation is triggered, matrix components can be eliminated within mitochondrion-derived vesicles (MDVs) budding from dysfunctional but not yet depolarized mitochondria. 75,76 MDVs serve to eliminate oxidized mitochondrial elements through PTEN-induced putative kinase 1 (PINK1) and the mitochondrial (PINK1)-E3 ubiquitin-protein ligase Parkin. 77 The delivery of damaged cargo within MDVs to lysosomes is part of MQC and occurs as an early response to oxidative stress. 75 –77 Conversely, mitochondria that are severely damaged are fissioned and targeted for elimination through a distinct pathway involving the synergistic activity of the mitochondrial dynamics machinery, PINK1, Parkin, Bnip3L/Nix, and Bnip3. 78
General autophagy and mitophagy can control inflammation by either clearing apoptotic corpses through macrophage activity or inhibiting NLRP3 inflammasome activation. The inhibition of mitophagy results in spontaneous inflammasome activation as a consequence of mitochondrial ROS burst. 52,79 –81 Accordingly, autophagy-deficient cells show accumulation of abnormal mitochondria with increased levels of ROS and reduced membrane potential. 52 Recently, the activation of caspase-1 by NLRP3 has been shown to block mitophagy, thus reducing the clearance of damaged mitochondria and, as a positive feedback response, enhancing inflammasome activation. 82 This and other findings strongly support the existence of a pathway in which NLRP3 responds to mitochondrial dysfunction (reviewed in Gurung et al. 83 ). More specifically, the accumulation of severely damaged mitochondria due to defective mitophagy could result in the extrusion of components able to induce NLRP3-mediated inflammation (Fig. 1).
Cell death has been suggested to be another modulator of immune response through mitochondrial involvement and inflammasome activation. 51,84 Adenosine triphosphate (ATP) release following cell death has been proposed to function as a danger signal implicated in NLRP3 inflammasome activation alerting circulating neutrophils. 49 Interestingly, this pathway requires bioenergetically competent mitochondria, as shown by the inhibition of ATP-induced NLRP3 inflammasome activation when mitochondria are uncoupled. 85 He at al. 86 recently proposed that NLRP3 inflammasome activation by ATP in macrophages occurs through a double-step process. According to this view, NLRP3 expression activation is primed by microbial products or inflammatory cytokines via NF-κB signaling induction, while the actual inflammasome activation is triggered by ATP, pore-forming toxins, viral RNA, or particulate matter. 86
Moreover, alterations of mtDNA and TFAM content and expression of autophagy-related genes have been found in several mitophagy dysfunction-associated conditions. Only recently, the modulation of TFAM expression, TFAM-binding to mtDNA and mtDNA content has been proposed as a mechanism regulating mitochondrial biogenesis in tissues of aged rats on calorie restriction. 87 Oxidative modifications occurring at the level of TFAM or mtDNA are indicated as major factors affecting TFAM binding and resulting in nucleoid instability. The recent identification of a hotspot for oxidative modifications within the D-Loop region of the mitochondrial genome at the site of TFAM binding further supports the existence of an oxidative-related modulation of both replication and transcription. 88 Both cell-free mtDNA and TFAM-bound mtDNA have been shown to act as specific DAMPs eliciting a systemic inflammatory response. 14 Accordingly, oxidized HMGB1, a member of HMG box family of DNA, released by necrotic cells, has been shown to trigger a powerful immune response. 47,89 A potential role for oxidized TFAM both in the modulation of its binding to mtDNA and the inflammatory response associated with muscle wasting syndromes represents an uninvestigated interesting scenario.
Mitochondrial Dysfunction in Muscle Wasting Syndromes: A Focus on Sarcopenia and Cachexia
Muscle wasting is a feature of several conditions (e.g., sarcopenia, cachexia, neuromuscular disorders, diabetes) characterized by functional impairment and physical disability. The maintenance of mitochondrial function is especially relevant to myocyte viability because of their high reliance on oxidative metabolism for energy production. Due to their postmitotic nature, skeletal myocytes cannot clear damaged organelles through cellular division, but depend on the efficiency of quality control processes to preserve mitochondrial homeostasis. Therefore, derangements at any level of the MQC axis can easily result in mitochondrial dysfunction and energy deficiency. 90 Indeed, damaged mitochondria accumulate in myocytes of transgenic mice with abrogation of autophagy, inducing oxidative stress, apoptosis, and eventually muscle atrophy and weakness. 91,92
Sarcopenia, the progressive age-related decline in muscle mass and strength/function, is a complex and multifaceted process associated with an increased risk of adverse health outcomes (e.g., disability, loss of independence, morbidity, mortality). 93 Among the possible pathogenic mechanisms of sarcopenia, mitochondrial dysfunction has been actively investigated. 94 –100 Although the causes of mitochondrial damage during muscle aging are not completely understood, impairments in MQC with defective autophagy are thought to play a significant role. 100 –104
Overactivation of autophagy, on the other hand, has been proposed as a mechanism in cachexia, a multifactorial syndrome secondary to specific disease conditions, characterized by loss of body weight due to skeletal muscle wasting with or without adipose tissue depletion. 105 Several proinflammatory cytokines (e.g., TNF-α, IL1β, IL6, TNF-like weak inducer of apoptosis [TWEAK]), which are able to induce muscle catabolism, are elevated during cachexia (reviewed in Zhou et al. 106 ). Recent findings by our group also suggest that, in muscle of gastric cancer patients with cachexia, the MQC axis may be compromised at several checkpoints, including mitochondrial dynamics, tagging for disposal, and execution of mitophagy. 107
The existence of crosstalk between inflammation and dysfunctional autophagy in muscle wasting represents a novel and potentially relevant field of research. Indeed, mitochondrial impairment and systemic inflammation are hallmarks of both cachexia and sarcopenia. To the best of our knowledge, only one study focused on the interface between chronic inflammation and mitochondrial clearance in skeletal muscle in the context of aging and physical frailty. 108 This investigation made use of IL10-null mice (IL-10tm/tm), a rodent model of chronic inflammation and frailty, and reported severe mitochondrial damage with disrupted organelle ultrastructure and abnormal autophagomes in skeletal muscle. 108 Although these findings suggest the existence of a connection among mitochondrial dysfunction, cellular quality control failure, and inflammation, the signaling pathways responsible for such a link are yet to be elucidated. Circulating mtDNA, either in the form of cell-free “naked” or TFAM-bound molecule, is a prominent candidate for this role, being an important DAMP associated with inflammation and arising directly from mitochondrial damage. 18 Candidate triggers of inflammation in sarcopenia and cachexia could be represented by oxidized cell free-mtDNA or nucleoids extruded from damaged mitochondria (Fig. 2). These DAMPs, in turn, would interact with PRRs, leading to activation of innate immune system and subsequent production of inflammatory mediators. These latter could sustain a vicious circle in myocytes with impaired MQC, resulting in further mitochondrial impairment, increased ROS generation, and release of DAMPs-enriched MDVs. This series of events would favor the propagation of sterile inflammation, ultimately contributing to muscle wasting.
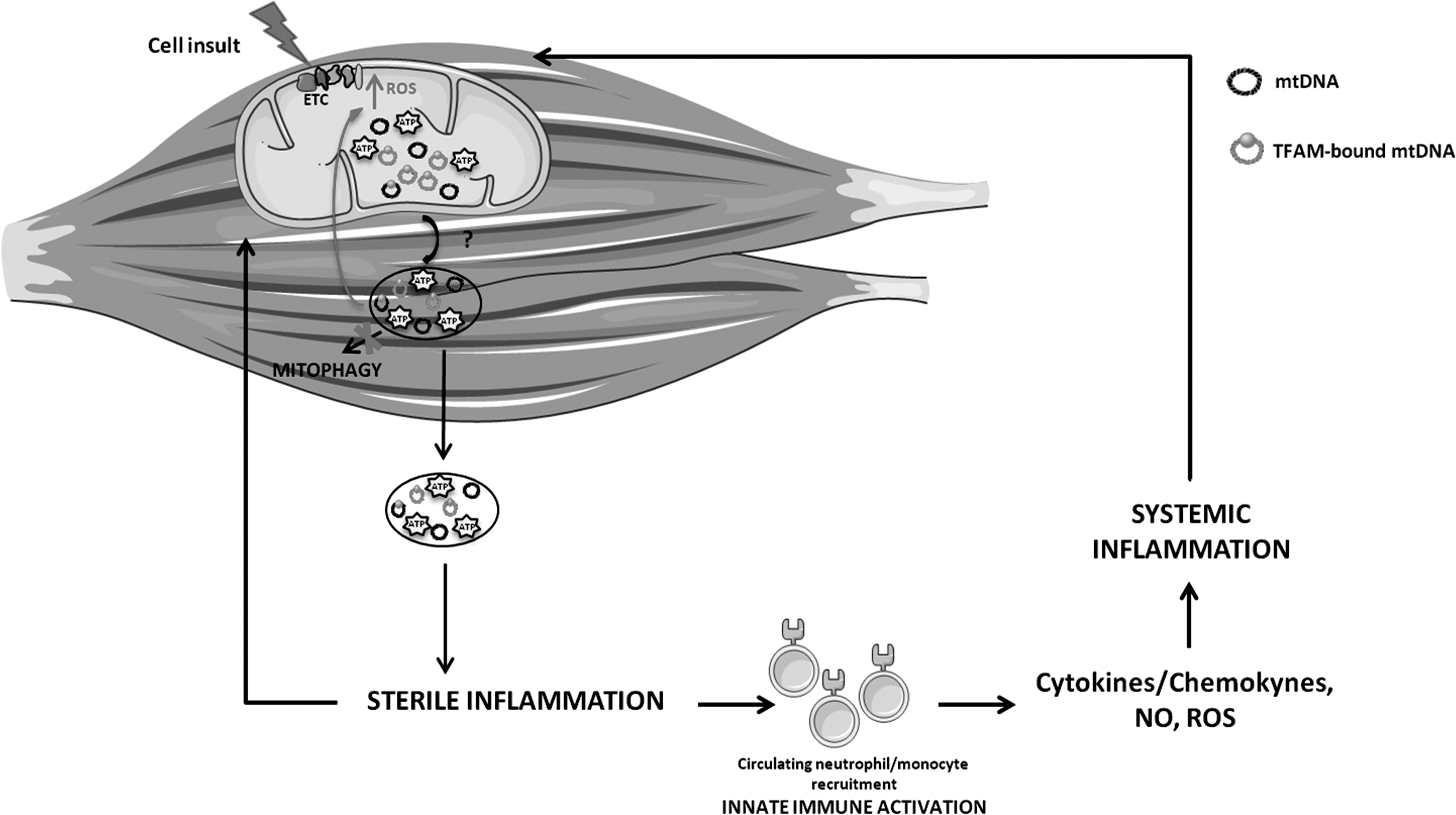
Proposed crosstalk between mitochondrial dysfunction and “sterile” inflammation during muscle wasting. The impairment of mitochondrial quality control in myocytes may lead to the release of mitochondrial damaged-associated molecular patterns, such as mtDNA and ATP, and subsequent recruitment of local macrophages. A persistent inflammatory trigger may also alert circulating immune cells, which, in turn, may mount a systemic response through the activation of mtDNA-induced inflammatory pathways. Cytokines, chemokines, NO, and ROS, released in the circulation by inflammatory cells, can induce further mitochondrial damage, thereby establishing a vicious circle and eventually contributing to muscle wasting. ATP, adenosine triphosphate; ETC, electron transport chain; NO, nitric oxide; TFAM, mitochondrial transcription factor A.
Conclusions and Future Perspectives
Sterile inflammation has received increasing attention due to its implication in the aging process and age-associated conditions. The intimate mechanisms underlying inflamm-aging are not fully elucidated. Yet, a danger cellular-driven response, fueled by DAMPs originating from dysfunctional mitochondria, is proposed to play an important role in senescence and chronic degenerative diseases.
Inflammation and mitochondrial dysfunction have a great impact on the development and progression of several muscle wasting disorders, including sarcopenia and cachexia. The identification of specific “danger molecules” that stimulate sterile inflammation and link this process with muscular mitochondrial dysfunction would therefore expand our understanding of muscle wasting pathophysiology. Such a knowledge could also lead to unraveling new signaling pathways for the design of targeted interventions. Given the importance of mitochondrial homeostasis to muscle health, the role played by mitochondrial DAMPs (e.g., mtDNA either unbound or in association with TFAM), which arise from organelle damage and failing MQC, represents a relevant matter to be addressed by future investigations.
Footnotes
Acknowledgments
This work was supported by Fondazione Roma (NCDs Call for Proposals 2013), Innovative Medicine Initiative-Joint Undertaking (IMI-JU No. 115621), intramural research grants from the Catholic University of the Sacred Heart (D3.2 2013 and D3.2 2015), the nonprofit research foundation “Centro Studi Achille e Linda Lorenzon,” and the Claude D. Pepper Older Americans Independence Center at the University of Florida's Institute on Aging (NIA 1P30AG028740).
Author Disclosure Statement
E.M., F.L., R.B., and R.C. are partners of the SPRINTT consortium, which is partly funded by the European Federation of Pharmaceutical Industries and Associations (EFPIA). E.M. served as a consultant for Huron Consulting Group, Genactis, Novartis, and Nutricia. R.C. served as a consultant from Novartis and Nutricia. All other authors have no competing financial interests to declare.