
Abstract
Electroacupuncture (EA) pretreatment induces cerebral ischemic tolerance; however, the mechanism remains poorly understood. This study aimed to determine the participation of peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α)-mediated mitochondrial biogenesis in the neuroprotection of EA and whether cannabinoid receptor 1 (CB1R) is involved in this mechanism. At 2 hours after EA pretreatment, adult male C57BL/6j mice were subjected to 60-minute right middle cerebral artery occlusion (MCAO). Mitochondrial function, the level of mitochondrial biogenesis-related proteins (nuclear transcription factor 1, NRF1; mitochondrial transcription factor A, TFAM), and mitochondrial DNA (mtDNA) were measured. A small interfering RNA (siRNA) targeting PGC-1α and the CB1R antagonists AM251 and SR141716A were given to the animals before EA pretreatment, and mitochondrial function and biogenesis were examined after MCAO. EA ameliorated the mitochondrial function, upregulated the NRF1 and TFAM expression, and increased the mtDNA levels and the volume and number of mitochondria. EA pretreatment increased the expression of PGC-1α, whereas the PGC-1α siRNA and CB1R antagonists reversed the improved neuroprotection and increased mitochondrial biogenesis induced by EA. Our results indicated that EA pretreatment protects the mitochondria and promotes mitochondrial biogenesis by activating CB1R-dependent PGC-1α, which provides a novel mechanism for EA pretreatment-induced ischemic tolerance.
Introduction
Stroke, mainly ischemic stroke, is still one of the leading causes of disability and mortality in China. 1 Although tissue plasminogen activator (t-PA) has been approved by the Food and Drug Administration for stroke treatment, the narrow therapeutic window (<4.5 hours) and safety concerns limit the use of this treatment, and no more than 5% of patients can be benefited from t-PA. 2 Thus, new effective medications or strategies for stroke treatment are urgently needed. In studies of stroke therapy, electroacupuncture (EA) stimulation at the “Baihui (GV20)” acupoint for 30 minutes (EA pretreatment), a treatment derived from traditional Chinese medicine, is one of the most commonly proposed pretreatment strategies for experimental ischemic stroke. 3,4 However, the exact molecular and cellular mechanism underlying the neuroprotection induced by EA pretreatment is still unclear.
During various physiological processes, mitochondria are crucial in generation of reactive oxygen species (ROS), maintenance of iron homeostasis, and regulation of programmed cell death signaling. Mitochondrial dysfunction has been related to the pathogenesis of ischemic/reperfusion (I/R) injury. 5,6 After I/R challenge, mitochondrial dysfunction leads to the failure of the energy supply to cells, excess ROS generation, and the activation of cytochrome C-regulated mitochondrial apoptotic cascades, which in turn reinforce the injury. 7,8 As highly dynamic organelles, mitochondria continually undergo fission (mitochondrial biogenesis) and fusion (mitochondrial autophagy or mitophagy). Growing evidence has demonstrated that mitochondrial dynamics, including mitochondrial biogenesis, play a determinant role in the maintenance of mitochondrial function and the process of cerebral I/R injury. 9,10 In the regulation of mitochondrial biogenesis, peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) has been determined to be an emerging stimulator of mitochondrial biogenesis. 11 In the central nervous system, the activation or overexpression of PGC-1α has been shown to upregulate mitochondrial biogenesis, reduce mitochondrial loss, and ameliorate mitochondrial dysfunction. 11 –14 However, whether EA pretreatment exerts a neuroprotective effect through the promotion of PGC-1α-dependent mitochondrial biogenesis is still unclear.
Cannabinoid receptor 1 (CB1R), a G protein-coupled receptor that is centrally located in the neuronal plasma membrane, is involved in modulation of neuronal activity, synaptic plasticity, and cell metabolism. 15,16 Accumulating evidence, including our findings, has demonstrated that CB1R-mediated protective signaling pathways are involved in the neuroprotective effect induced by EA pretreatment. 3,17 –20 Some studies have also indicated that the CB1 receptor participates in mitochondrial biogenesis in non-neuronal tissues. 21,22 Moreover, exogenous supplementation of marijuana-derivative cannabis Δ9-tetrahydrocannabinol or the highly selective CB1R agonist arachidony1-2-chloroethylamide (ACEA) induced neuroprotective effects by enhancing mitochondrial biogenesis and maintaining mitochondrial function. 23,24 However, to our knowledge, whether EA pretreatment promotes mitochondrial biogenesis through a CB1R-dependent mechanism is still unknown.
In this study, we used a transient focal cerebral I/R model in mice to investigate the role of CB1R-induced activation of PGC-1α in the enhanced mitochondrial biogenesis and the neuroprotection induced by EA pretreatment.
Materials and Methods
Animals
All animal-related procedures were approved by the Ethics Committee for Animal Experimentation of Fourth Military Medical University (Xi'an, China) and preceded in accordance with the National Institutes of Health Guide for the Care. A randomized number table was used for randomization in this study. Analgesia was applied with the administration of meloxicam (i.p.) at 0.2 mg/kg body weight postoperatively and was followed by administration of 0.05 mg/kg body weight for 3 consecutive days. Male C57BL6j mice between 8 and 10 weeks old (25–30 g) were purchased from the Animal Laboratory of the Fourth Military Medical University, Xi'an, China. The mice were maintained in a 12-hour alternating light and dark cycle at 20–25°C and 60–70% humidity with freely available water and food for at least 1 week before treatment or surgery. The number of animals used and their suffering were minimized in this study.
EA pretreatment
EA pretreatment was performed as described in our previous studies. 4,17 Hence, the acupoint Baihui (GV20) was stimulated with an intensity of 1 mA and a frequency of 2/15 Hz for 30 minutes using the Hwato EA Instrument (Model No. SDZ-V, Suzhou Medical Appliances Co., Ltd., Suzhou, China). Mice were subjected to middle cerebral artery occlusion (MCAO) or a sham operation 2 hours after EA stimulation.
Drug and siRNA administration
Two different CB1R antagonists, AM251 and SR141716A, were also administered based on previous reports. 17 In brief, the CB1R antagonists AM251 and SR141716A were dissolved in dimethyl sulfoxide (DMSO) and Tween-80, respectively, followed by dilution in saline. The proportion of DMSO/Tween-80/saline was 1:1:18. Based on a protocol used in a previous study, a dosage of 1 mg/kg for both antagonists was given 30 minutes before the EA pretreatment.
PGC-1α small interfering RNA (PGC-1α-siRNA) and its negative control (PGC-1α-siRNAc) were purchased from GeneChem Co., Ltd. (Shanghai, China). The target sequences were as follows: sense primer 5′-GCUCUUGAGAAUGGAUAUATT-3′ and antisense primer 5′-UAUAUCCAUUCUCAAGAGCTT-3′. PGC-1α-siRNA or PGC-1α-siRNAc transfection was performed by intracerebroventricular injection. The stereotaxic coordinate location of the lateral cerebral ventricle was 0.4 mm posterior to the bregma, 1.0 mm lateral to the midsagittal line, and 2.0 mm deep from the cranial surface. 25 At 72 hours after surgery, the efficacy of the PGC-1α-siRNA or PGC-1α-siRNAc was determined by western blot analysis.
Establishment of transient focal cerebral I/R injury
Focal cerebral I/R injury was induced by MCAO according to a protocol described previously. 17 In brief, mice were anesthetized with 10% chloral hydrate (350 mg/kg, i.p.) after an overnight fast. After the right carotid arteries were exposed, an intraluminal 6-0 nylon monofilament with a round tip was inserted from the right common carotid artery to the right middle cerebral artery, and then, the filament was withdrawn to allow reperfusion after 1 hour for occlusion. During the surgery, the temporal temperature was maintained at 37°C ± 0.5°C by a thermostatic blanket and a lamp. For monitoring the regional cerebral blood flow (rCBF), a laser Doppler sensor was placed on the surface of the skull 2 mm caudal and 4 mm lateral to the bregma. Only the mice with 80% drop during occlusion and 70% recovery during reperfusion in the rCBF were allowed to the next experiments. For mice administrated with PGC-1α-siRNA or PGC-1α-siRNAc, MCAO was performed 72 hours after microinjection.
For the sham operation, the mice were subjected to the same surgical procedure without inserting filaments. After recovery from anesthesia, the mice were returned to their cages with freely available water and food.
Evaluation of neurological score and infarct volume
The neurological behavioral outcome in mice was evaluated with the Garcia Score Scale by an observer who was blinded to all animal group settings. 26 The infarct volume was measured by 2,3,5-triphenyltetrazolium chloride (TTC, 1%; Sigma-Aldrich, St. Louis, MO) staining. After the behavior test, the mice were killed, and the brain was rapidly transferred to ice-cold saline for 5 minutes and then used to generate 1-mm-thick coronal sections. The slices were incubated in TTC for 10 minutes and then fixed with 4% paraformaldehyde in 0.01 M PBS (pH 7.4) for 24 hours. Next, the slices were photographed, and the infarct size was calculated according to Swanson's method to correct for edema: 100% (contralateral hemisphere volume − nonlesioned ipsilateral hemisphere volume)/contralateral hemisphere volume. 27
Western blot analysis
At 4 and 24 hours after reperfusion, the mice from each group were killed, and brain tissue from the penumbra area was harvested. Proteins were extracted from homogenized tissue in ice-cold RIPA lysis buffer containing 1% phenylmethanesulfonyl fluoride (Beyotime, Nantong, China). Preparation of cytosolic protein was achieved with a commercially available cytosol/mitochondria fractionation kit (Beyotime Biotechnology, China) in accordance with the manufacturer's paradigm. The protein concentration was determined by the Bradford method according to the manufacturer's instructions. Then, 30 μL of protein was loaded onto polyacrylamide–sodium dodecyl sulfate gels. Western blot analysis was performed as reported previously. The primary antibodies used in this study were as follows: cytochrome C (Abcam; 1:1000 dilution), COX IV (Abcam; 1:1000 dilution), NRF-1 (Santa Cruz; 1:500 dilution), TFAM (Santa Cruz; 1:500 dilution), PGC-1α (Cell Signaling Technology; 1:500 dilution), and GAPDH (Kangwei; 1:1000 dilution). After the membranes were washed three times in Tris-buffered saline containing Tween-20, they were incubated with secondary horseradish-conjugated goat anti-rabbit secondary antibody at room temperature for 1 hour. The quantitative analysis for all the specific protein bands was performed after scanning by enhanced chemiluminescent reagent (ECL; Millipore). All changes in protein expression were normalized to the ratio of GAPDH.
Double immunofluorescence staining
Brains were cut on a cryostat into 10-μm-thick coronal sections at 1.3 mm rostral from the bregma. For immunofluorescence staining, the sections were washed three times with phosphate-buffered saline containing 1% Triton and then incubated with neuronal nuclei antibody (1:1000; Millipore) and PGC-1α (Cell Signaling Technology; 1:200 dilution) for 24 hours at 4°C. After incubation, all slices were washed with PBS three times and then incubated with CY3-labeled goat anti-rabbit and FITC-labeled goat anti-rat secondary antibodies (1:1000 for both; Millipore) at room temperature for 2 hours. Next, the slices were incubated with the nuclear marker DAPI (1 ng/μL; Sigma) for 5 minutes at room temperature. The fluorescent signals from penumbra area were determined using a confocal fluorescence microscope (Flv100i; Olympus).
TUNEL staining for analysis of apoptosis
Terminal deoxynucleotidyl transferase deoxyuridine triphosphate-biotin nick-end labeling (TUNEL) staining was used to evaluate neuronal apoptosis 72 hours after reperfusion. With the in situ cell death detection kit (Roche, Germany), TUNEL staining with co-labeling of a neuronal marker in neuron nuclei was performed according to a standard protocol. The TUNEL-positive neurons were acquired using a 40 × objective lens from areas in the ischemic penumbra, and the number of TUNEL-positive cells is expressed as the number per 100 br 2 . The ischemic penumbra area was defined as reported previously.
RNA isolation and real-time RT-PCR of mitochondrial DNA
Total RNA was isolated from the harvested frozen penumbra using TRIzol according to the manufacturer's instructions. Each 2–3 μg template RNA was used to synthesize cDNA using a reverse transcription kit (TaKaRa).
Real-time PCR of mitochondrial DNA (mtDNA) was performed using the forward and reverse primer sequences as follows: forward primer: ATATTTTCACTGCTGAGTCCCGTGG, reverse primer: AATTTCGGTTGGGGTGACCTCGGAG. The data analysis was performed with a comparative critical threshold (Ct) method where the amount of target was normalized to the amount of endogenous control.
Assessment of mitochondrial function in isolated mitochondria
Mice were killed after deep anesthetization with chloral hydrate, and the penumbra tissues were dissected as described previously. 28 Mitochondrial isolation was performed using a proteome mitochondria isolation kit (Pierce; 89874) in compliance with the manufacturer's instructions. The concentration of mitochondrial protein was examined by a Bradford assay kit (Beyotime, Nantong, China) with bovine serum albumin as a standard. All isolated mitochondria were diluted with swelling buffer (KCl, 120 mM; Tris-HCl, 10 mM; MOPS, 20 mM; KH2PO4, 5 mM; at pH 7.4) to a final concentration of 0.5 mg protein/mL. All experiments with isolated mitochondria were performed within 3 hours.
Measurement of mitochondrial radical production
The total radical production in the isolated mitochondria was measured by a kit (Beyotime) according to our previously reported protocol. 29 In brief, 10 μL of isolated mitochondria was suspended in 170 μL of HEPES buffer to a concentration of 10 mM in 96-well cluster plates. Then, 20 μL of 100 mM DCFH-DA was added to each well at a final volume of 200 μL, and the plate was placed in an incubator at 30°C for 30 minutes. After incubation, the fluorescence of each well was measured using a Cary Eclipse Fluorescence Spectrometer. The excitation and emission were set at 485 and 528 nm, respectively.
Assessment of mitochondrial permeability transition pore opening
The examination of the mitochondrial permeability transition pore (mPTP) was performed as described previously by the calcein–cobalt method with an available kit. 30 In brief, when mitochondria swell and the mPTP opens, calcein is released into the cytoplasm and can be measured by a spectrophotometer, which is used as an indicator of MPTP. The fluorescence intensity was measured with a monochromator microplate reader using excitation at 488 nm and emission at 505 nm.
Measurement of citrate synthase and mitochondrial membrane potential
Citrate synthase activity was examined in isolated mitochondria according to previous studies using an available kit according to the manufacturer's instructions (Sigma). 30 Hence, the examination was confirmed in 10 μg mitochondria samples following the reduction of 5,5′-dithiobis (2-nitrobenzoic acid) at 412 nm, which was normalized to the attenuation of coenzyme A by the citrate synthase reaction in the presence of oxaloacetate.
The fluorescent probe JC-1 (5,5,6,6-tetrachloro-1,1,3,3-tetraethylbenzimidazolcarbocyanine iodide) was used to measure MMP with a kit according to the method reported previously (Sigma). 31 In brief, 0.5 mg/mL mitochondrial samples (1 mL total) were incubated with 19 mL of JC-1 staining buffer. The fluorescence intensity was examined at 37°C in a fluorescence spectrophotometer. The ratio between aggregates (red, 590 nm) and monomers (green, 525 nm) was calculated. In this study, valinomycin was applied as the negative control, and the results were corrected for mitochondrial viability (release of cytochrome C oxidase; Abcam) and are expressed as a percentage.
Examination of mitochondrial volume and number
The structure of the mitochondria in the penumbra area was assessed by transmission electron microscopy (TEM) 4 hours after reperfusion. In brief, vibratome-prepared sections were selected and transferred into phosphate buffer. Then, the tissues were rinsed in buffer and post-fixed with 1% osmium tetroxide for 1 hour and subsequently transformed to a graded ethanol dehydration and infiltrated with a 50:50 mixture of propylene oxide and resin for an overnight incubation. Seventy-two hours later, the tissues were embedded in resin and cut into 60 nm sections. The sections were stained with 2% uranyl acetate for 20 minutes and 0.5% lead citrate for 5 minutes. The ultrastructure was visualized using a transmission electron microscope (Philips, the Netherlands). For the morphometric analysis of mitochondria, 15 areas including large neuronal-like nuclei covering approximately one-fourth of the visible image were selected per mouse. All images were photographed at × 30,000 magnification and counted (three mice per group). The observation and analysis of mitochondrial volume and number were confirmed by an operator who was blinded to all groups.
Statistical analysis
For data analysis, the software SPSS 18.0 for Windows was used to conduct the statistical analyses. Neurological scores are expressed as the median with the interquartile range and were analyzed by the Kruskal–Wallis test followed by the Mann–Whitney U test and a Bonferroni post hoc test. The other data are represented as mean ± SEM and were analyzed by one-way analysis of variance with a Bonferroni correction for post hoc correction. Two-tailed values of p < 0.05 were considered statistically significant.
Results
EA pretreatment alleviated the mitochondrial dysfunction after cerebral ischemia injury
As given in Figure 1B, the release of cytochrome C in cytoplasm was reduced in the EA + MCAO group compared with the MCAO group at 24 hours after reperfusion (p < 0.05). The COX IV protein level (Fig. 1C) was upregulated in the EA pretreatment group (p < 0.05). To further determine the effect of EA on mitochondrial function, we examined the total radical production. As given in Figure 1D, elevated radical generation was detected in the mice subjected to MCAO 24 hours after reperfusion. This elevation was significantly reversed by EA pretreatment (p < 0.05). Compared with that of the sham group, the ΔA540 value was significantly increased at 24 hours after reperfusion in the MCAO group (p < 0.05; Fig. 1E), but this upregulation was reversed in the EA + MCAO group compared with the MCAO group (p < 0.05). The MMP level in the MCAO group was significantly attenuated compared with that in the sham group (p < 0.05; Fig. 1F). EA pretreatment significantly upregulated the MMP value compared with that in the MCAO group (p < 0.05). Furthermore, the value of citrate synthase in the MCAO group was lower than that in the sham group. When mice were pretreated with EA, the expression of citrate synthase was increased compared with that in the MCAO group (p < 0.05; Fig. 1G).

EA pretreatment alleviated the mitochondrial dysfunction induced by ischemia/reperfusion injury.
EA pretreatment promoted mitochondrial biogenesis after cerebral ischemia reperfusion
As given in Figure 2B and C, in animals pretreated with EA, the expression of NRF-1 and TFAM was significantly increased at 4 hours but not 24 hours after reperfusion compared with that of the MCAO group (p < 0.05). Moreover, the expression of mtDNA was increased in the EA group compared with the MCAO group (p < 0.05; Fig. 2D).

EA pretreatment increased mitochondrial biogenesis proteins and mitochondrial DNA at 4 hours after reperfusion.
EA pretreatment promoted the expression of PGC-1α and increased the number and volume of mitochondria after reperfusion
As given in Figure 3A, increased expression of PGC-1α was detected in the EA + MCAO group compared with the MCAO group at 4 hours but not 24 hours after reperfusion. As given in Figure 3B, the double immunofluorescence staining results indicated that the increased PGC-1α protein partly colocalized with neurons. The measurement of mitochondrial number and volume was performed by TEM at 4 hours after reperfusion (Fig. 3C). As given in Figure 3D, the mitochondrial volume was significantly increased in the EA + MCAO compared with the MCAO group (p < 0.05). Consistent with the increased mitochondrial volume, EA pretreatment also increased the number of mitochondria in the penumbra area at 4 hours after reperfusion compared with that in the MCAO group (p < 0.05; Fig. 3E).

EA pretreatment increased the abundance of PGC-1α and changed the microstructure of the mitochondria.
The neuroprotective effect of EA pretreatment was absent in the mice administered PCG-1α-siRNA
As given in Figure 4B, the neurological score in the EA pretreatment group was significantly higher than that in the MCAO group at 72 hours after reperfusion. However, after PCG-1α-siRNA administration, the EA-pretreated mice showed a worse neurobehavioral performance than the EA and EA + PCG-1α-siRNAc mice at 72 hours after reperfusion (p < 0.05). When PCG-1α-siRNA was administered, the EA-pretreated mice had a larger brain infarct volume than the EA-pretreated mice (p < 0.05; Fig. 4C). No significant difference was found in a comparison with the EA or EA + PCG-1α-siRNAc group.

Supplementation with PGC-1α-siRNA reversed the neuroprotective effect of EA pretreatment.
Neuronal cell apoptosis in the ischemic penumbra was also detected by TUNEL immunofluorescence staining. As given in Figure 4D and E, at 72 hours after reperfusion, the number of TUNEL-positive neurons in the EA pretreatment group was significantly reduced compared with that in the MCAO group (p < 0.05). However, with PCG-1α-siRNA administration, the number of TUNEL-positive cells was significantly increased compared with that in the EA + PCG-1α-siRNAc group (p < 0.05).
Beneficial effect on mitochondrial function induced by EA pretreatment was abolished by the administration of PCG-1α-siRNA
As given in Figure 5B, the reduced release of cytochrome C in cytoplasm produced by EA pretreatment was reversed in the EA + MCAO + PCG-1α-siRNA group compared with the EA group (p < 0.05). No significant difference was detected between the EA and EA + MCAO + PCG-1α-siRNAc groups. The increased content of COX IV (Fig. 5C) in the EA pretreatment group was also reversed with the administration of PCG-1α-siRNA (p < 0.05). As given in Figure 5D, the reduced radical generation in the mice pretreated with PCG-1α-siRNA was significantly reversed by supplementation with PCG-1α-siRNA (p < 0.05). Compared with that of the EA + MCAO group, the ΔA540 value was significantly increased in the EA + MCAO + PCG-1α-siRNA group (p < 0.05, Fig. 5E), whereas this attenuation was not affected by treatment with PCG-1α-siRNAc. The MMP level in the EA + MCAO + PCG-1α-siRNA group was significantly reduced compared with that in the EA group (Fig. 5F, p < 0.05). Furthermore, the value of citrate synthase in the EA + MCAO + PCG-1α-siRNA group was higher than that in the EA + MCAO group (Fig. 5G, p < 0.05). When the mice were pretreated with PCG-1α-siRNAc, the expression of citrate synthase was not affected compared with that in the EA group (p > 0.05).

The ameliorated mitochondrial function induced by EA pretreatment was reversed by supplementation with PGC-1α-siRNA.
Supplementation with PCG-1α-siRNA ablated the increased NRF-1, TFAM, and mtDNA levels induced by EA pretreatment
As given in Figure 6B and C, in the EA animals pretreated with PCG-1α-siRNA, the expression of NRF-1 and TFAM was significantly reduced compared with that of mice in the EA group (p < 0.05). Moreover, the increased expression of mtDNA was reversed in the EA + MCAO + PGC-1α-siRNA group compared with the EA + MCAO group (Fig. 6D, p < 0.05). No significant difference was detected in NRF-1 and TFAM or mtDNA expression between the EA and EA + MCAO + PCG-1α-siRNAc groups.
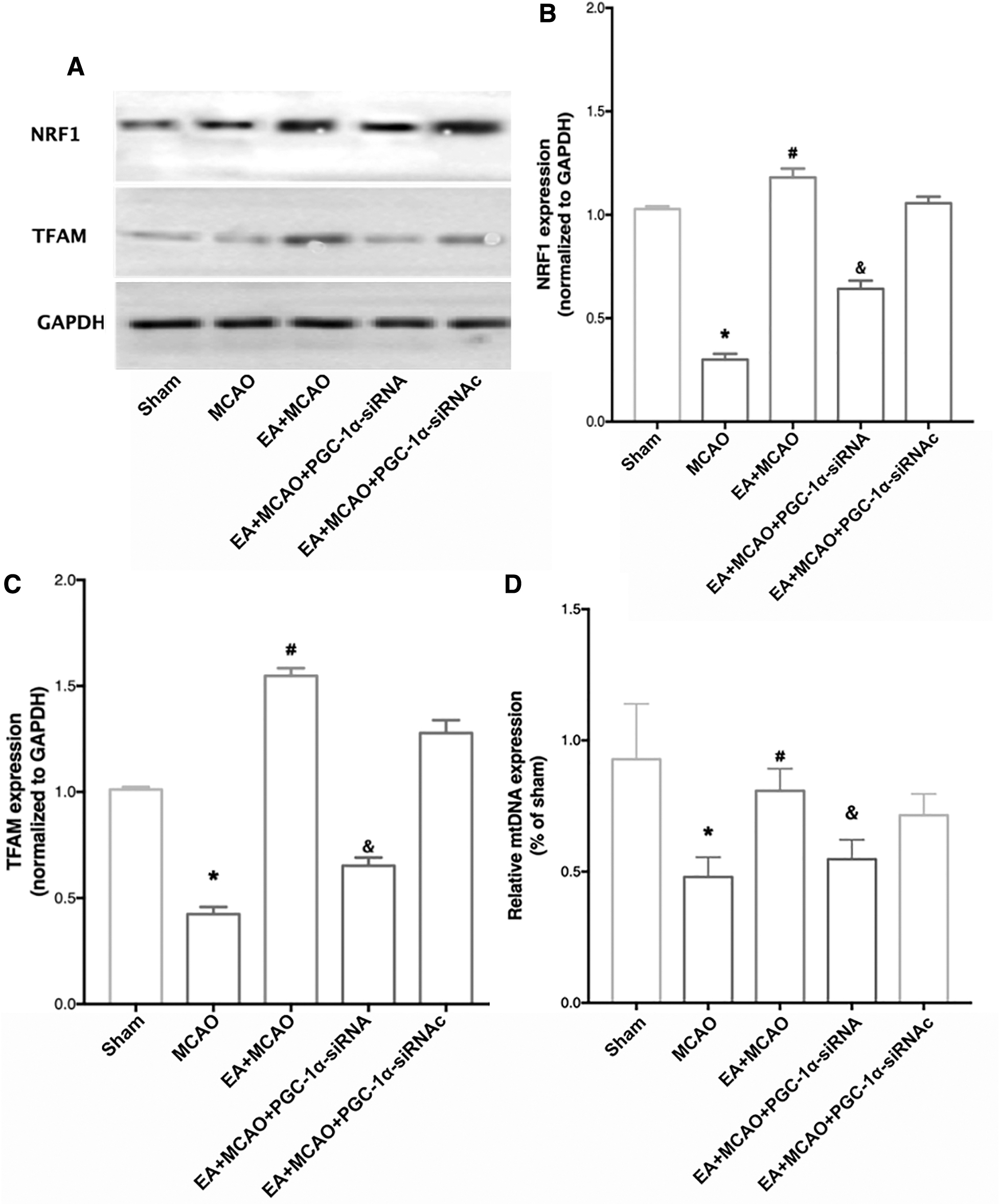
The increased mitochondrial biogenesis proteins and mitochondrial DNA induced by EA pretreatment were abolished by PGC-1α-siRNA administration.
Supplementation with the CB1R antagonists AM251 and SR141716A ablated the increased expression of PCG-1α and reversed the upregulated NRF-1, TFAM and mtDNA expression induced by EA pretreatment
As given in Figure 7A, the increased expression of PGC-1α detected in the EA pretreatment group was reversed by supplementation with the CB1R antagonists AM251 and SR141716A compared with that of the EA + MCAO + vehicle group (p < 0.05). As given in Figure 7B and C, the expression of NRF-1 and TFAM in the AM251- and SR141716A-treated mice was significantly reduced compared with that of mice in the EA + MCAO + vehicle group (p < 0.05). Moreover, the increased expression of mtDNA was reversed in the EA + MCAO + AM251 or EA +MCAO + SR141716A group compared with the EA + vehicle group (Fig. 7D, p < 0.05).
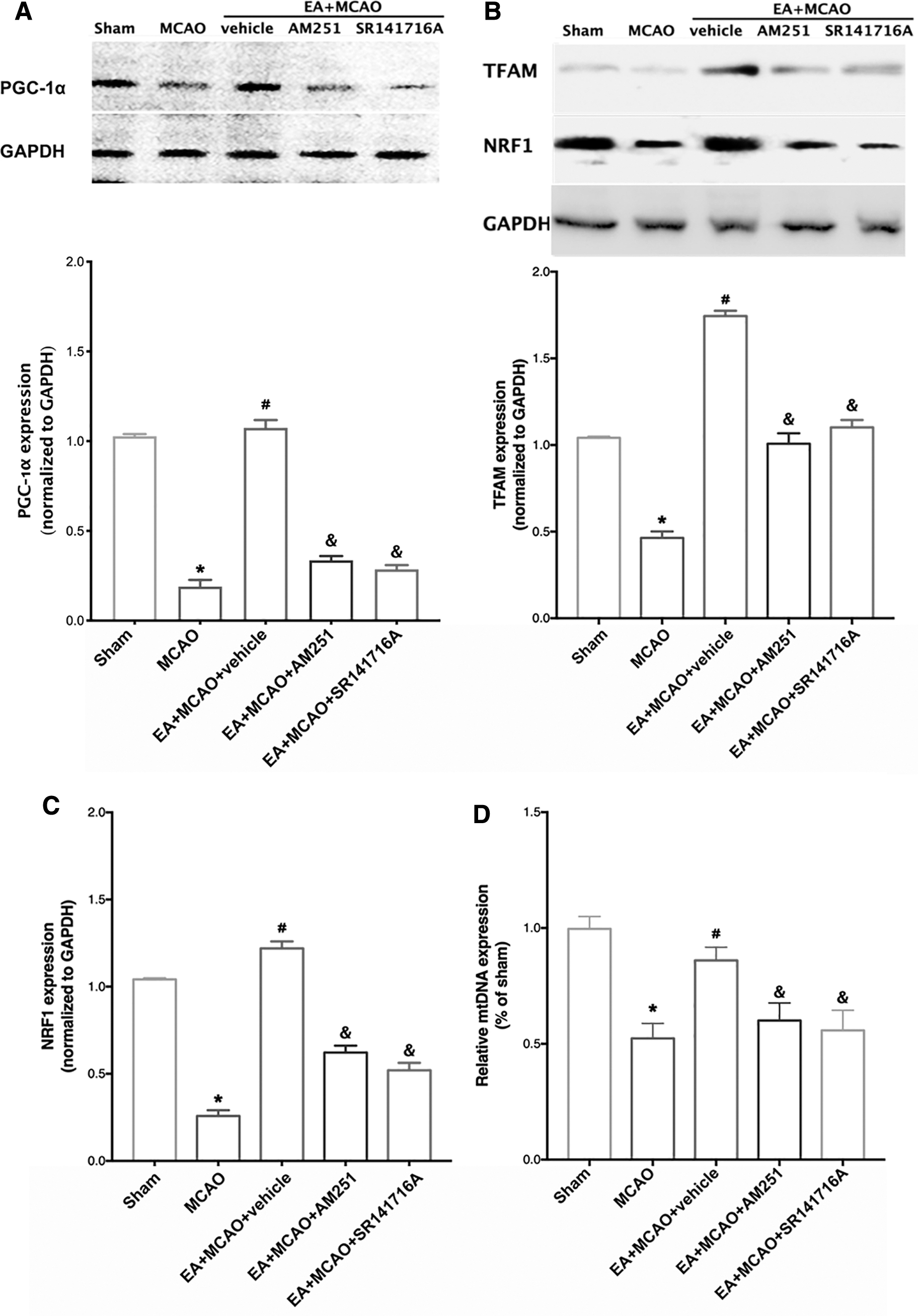
The CB1R antagonists AM251 and SR141716A abolished the upregulated expression of PGC-1α and the mitochondrial biogenesis proteins induced by EA pretreatment.
The ameliorated mitochondrial function induced by EA pretreatment was abolished in the mice treated with the CB1R antagonists AM251 and SR141716A
As given in Figure 8B, the reduced release of cytochrome C in cytoplasm induced by EA pretreatment was reversed in the EA + MCAO + AM251 and EA + MCAO + SR141716A group compared with the EA + vehicle group (p < 0.05). The increased content of COX IV (Fig. 8C) in the EA group was also reversed with the administration of AM251 or SR141716A (p < 0.05). As given in Figure 8D, the reduced radical generation in the mice pretreated with AM251 or SR141716A was significantly reversed (p < 0.05). Compared with that of the EA + vehicle group, the ΔA540 value was significantly increased in the EA + MCAO + AM251 and EA + MCAO + SR141716A groups (p < 0.05; Fig. 8E). The MMP level in the EA + MCAO + AM251 and EA + MCAO + SR141716A groups was significantly reduced compared with that in the EA + vehicle group (p < 0.05; Fig. 8F). In addition, either AM251 or SR141716A treatment reversed the increase in citrate synthase induced by EA pretreatment (Fig. 8G, p < 0.05).

The CB1R antagonists AM251 and SR141716A abolished the ameliorated mitochondrial function induced by EA pretreatment.
Discussion
This study explored the molecular mechanisms underlying the neuroprotective effect of EA pretreatment. Using a transient focal cerebral I/R model in mice, we demonstrated that CB1R-dependent PGC-1α activation induced by EA pretreatment participated in the promotion of mitochondrial biogenesis after brain ischemia. EA pretreatment ameliorated mitochondrial dysfunction, as indicated by the reduced cytochrome C release, the increased expression of COX IV, the reduced mitochondrial total free radical generation, the decreased MPTP opening, the hyperpolarized MMP, and the increased citrate synthase. These findings are relevant to the upregulated mitochondrial biogenesis, which was indicated by the upregulated expression of NRF1 and TFAM, increased mitochondrial numbers, and mtDNA expression. These benefits induced by EA pretreatment were reversed by supplementation with PGC-1α siRNA. Moreover, we found that CB1R was crucial for EA pretreatment-induced mitochondrial biogenesis. The findings of this study indicate a novel mechanism of EA pretreatment-induced neuroprotection against cerebral I/R injury.
As ubiquitous intracellular organelles, mitochondria are primarily responsible for various cellular contents and biological functions. Accumulating studies have shown that disruption of mitochondrial function plays a vital role in the pathogenesis of cerebral I/R injury. In addition, these findings have revealed the rationale for the improvement in mitochondrial function and the introduction of protective effects. 5,6 However, in the CNS, the mechanism underlying the regulatory effect of EA pretreatment on mitochondrial function is still unclear. Under ROS accumulation and Ca2+ overload induced by cerebral ischemia/reperfusion, mitochondrial membrane destabilization disturbs the proton gradient, which results in the opening of mPTP and the release of mitochondrial components such as cytochrome C to initiate the apoptotic cascade. Therefore, in this study, we first determined the effect of EA pretreatment on the amelioration of mitochondrial function, as indicated by the improved MMP depolarization, increased value of citrate synthase, attenuated mPTP opening, and decreased total radical generation. These results are consistent with our and other studies using different pretreatment stimuli. 29,32
Mitochondrial biogenesis is crucial for the maintenance of mitochondrial function. Recent advances have suggested an intimate correlation between mitochondrial biogenesis and neurological diseases, including ischemic stroke. 33 Furthermore, enhanced mitochondrial biogenesis has been found to augment cerebral tolerance against I/R insult and has been identified as a new target for therapeutic maneuvers to treat ischemic stroke. 5 Multiple protein factors have been implicated in the regulation of mitochondrial biogenesis. Nuclear respiratory factor (NRF1) plays a critical role in the control of the transcription of nuclear-encoded mitochondrial genes, which affect the electron transport chain and induce the expression of mitochondrial transcription factor A (TFAM). 34 TFAM has been shown to be crucial in the initiation of mtDNA replication and the transcription of mitochondrial-encoded genes. 35 These two proteins are generally accepted as indicators of mitochondrial biogenesis because of their major regulatory effect on mitochondrial biogenesis. 36 Consistent with other studies using cerebral hypoxic preconditioning that showed an increase in mitochondrial number, our study indicates that the neuroprotective effect of EA pretreatment is associated with increased mitochondrial numbers and upregulated levels of mtDNA and the mitochondrial biogenesis regulators NRF1 and TFAM. 37,38 Of note, increased expression of NRF1 and TFAM was not detected 24 hours after reperfusion, as was previously reported. This result indicated that the promoted mitochondrial biogenesis was an acute response to ischemic insult. 39
EA pretreatment protects the brain from I/R injury by promoting mitochondrial biogenesis; however, there is some debate regarding the nature of how EA produces mitochondrial biogenesis. Another main finding of this study is that EA pretreatment engages mitochondrial biogenesis through a signaling mechanism, as demonstrated by the capacity of EA to upregulate NRF1 and TFMA expression in a PGC-α-dependent manner in the heart, skeletal muscle, and in the brain. 40 –42 Previous evidence has indicated that the activation or overexpression of PGC-1α is a compensatory mechanism for neuronal mitochondrial loss. 43 –45 In this study, the PGC-1α protein expression was increased after EA pretreatment, as shown by western blot analysis and immunofluorescence staining. Moreover, some other studies have demonstrated the regulatory upstream mechanism of PGC-1α on NRF1 and TFAM. 46,47 To further clarify whether the activation of NRF1 and TFAM mediated by EA pretreatment is PGC-1α dependent, we used PGC-1α siRNA in this study. The silencing of PGC-1α resulted in the ablation of the upregulated expression of NRF1 and TFAM, ameliorated mitochondrial function and ischemic tolerance induced by EA pretreatment. Indeed, these findings correlate well with other investigations, indicating that the PGC-1α-NRF1-TFAM pathway is crucial for mitochondrial synthesis.
Another main finding of this study is that CB1R participates in PGC-1α-mediated stimulation of mitochondrial biogenesis. Extensive evidence has implicated CB1R and mitochondrial biogenesis in the peripheral and central nervous systems. 22 Thus, we applied the CB1R antagonists AM251 and SR141716A and found that these treatments abolished the EA pretreatment-mediated improvements in mitochondrial function. Consistent with previous studies, our studies also investigated that CB1R is involved in maintaining mitochondrial biogenesis. 23 The CB1R antagonists AM251 and SR141716A inhibited the expression of PGC-1α, indicating that, under EA pretreatment, CB1R-induced mitochondrial biogenesis occurs through the upregulation of PGC-1α.
However, some limitations of this study should be noted. First, we did not use selective techniques to directly inhibit mitochondrial biogenesis; thus, we could not conclude that mitochondrial biogenesis has a necessary role in the neuroprotection induced by EA pretreatment. In addition, we tried to investigate the role of PGC-1α expression in EA pretreatment; some studies have determined that the phosphorylation, ubiquitination, and acetylation of PGC-1α are also important in the activity of this protein, and whether these different regulatory mechanisms of PGC-1α participate in this process should be explored. Moreover, recent advance indicated some technique such as in vivo two-photon microscopy imaging could specifically track mitochondria in neurons; we would like to apply this new method in our future studies.
In summary, this study demonstrated that CB1R is involved in EA pretreatment-promoted mitochondrial biogenesis and induced cerebral ischemic tolerance by improving the expression of PGC-1α after cerebral I/R. These findings highlight a novel mechanism of EA pretreatment-induced neuroprotection.
Footnotes
Authors' Contribution
S.S., M.C., and Q.W. were involved in the design and conduct of the study, collection and analysis of the data and writing the article. T.J., N.D., M.W., and C.Y. participated in the study, data collection, and analysis. Y.L. and M. C. helped with the study design, data analysis, and completion of the article. Q.W. and M.C. supervised the study. All authors approved the final version of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from the National Natural Science Foundation of China (No. 81701145 to M.C., No. 81603408 to S.S., and No. 81974540 to Q.W.), and the Natural Science Basic Research Program of Shaanxi (Program No. 2017JZ029).