
Abstract
SUMOylation, a conserved protein post-translational modification that performs multiple functions including regulation of nuclear transport and transcription, is implicated in numerous biological processes including aging. RNAi knockdown of the sole Small Ubiquitin-like MOdifier (SUMO) gene, smo-1, in Caenorhabditis elegans shortened lifespan, whereas overexpression in the intestine modestly increased lifespan. Smo-1 is required for mitochondrial fission in a tissue-specific manner. Fission, in turn, is needed for mitophagy to maintain mitochondrial homeostasis during aging. SUMOlyation affects DAuer Formation (DAF)-16, which can be directly SUMOylated, and SKN-1, the homolog of mammalian Nrf2. These regulators play key roles in maintaining mitochondrial homeostasis. However, given the modest effect of overexpressing smo-1 on lifespan enhancement and potential interference with other genes that can promote increased lifespan, caution is advised in the translation of this study based on C. elegans. Although inhibitors of SUMOlyation have been developed for cancer and activators also have been identified, broad-acting biochemical pathway modifiers such as SUMO are often suboptimal drug targets and may not be as promising for antiaging applications as they first appear.
SUMOylation
Like ubiquitination, SUMOylation involves the post-translational modification of a target protein (Fig. 1). In this case, a series of enzymes attach a Small Ubiquitin-like MOdifier (SUMO) to the target protein as first reported by Mahajan et al. 1 Although ubiquitin attachment tags a target protein for proteasome degradation, SUMOylated proteins play critical regulatory roles in many cellular processes. 2,3 Hundreds of proteins regulating chromatin organization, transcription, DNA repair, macromolecular assembly and protein homeostasis, nuclear trafficking, and signal transduction are subject to reversible SUMOylation. Of these, transportation of SUMOylated proteins from the cytoplasm to the nucleus and transcription factor inactivation are among the most important. There are four confirmed SUMO isoforms in humans, each comprising ∼100 amino acids. SUMO-1 is ∼50% identical with the nearly identical SUMO-⅔ proteins. The consensus SUMOylation signal is a tetrapepide: Ψ-K-x-D/E, where Ψ is a hydrophobic amino acid, K is lysine (target of the SUMO conjugation), x is any amino acid, and D or E is an acidic residue (asp or glu). Princz et al. characterized SUMOylation in model organism Caenorhabditis elegans finding that SUMO plays a role in determination of lifespan and health span. 4
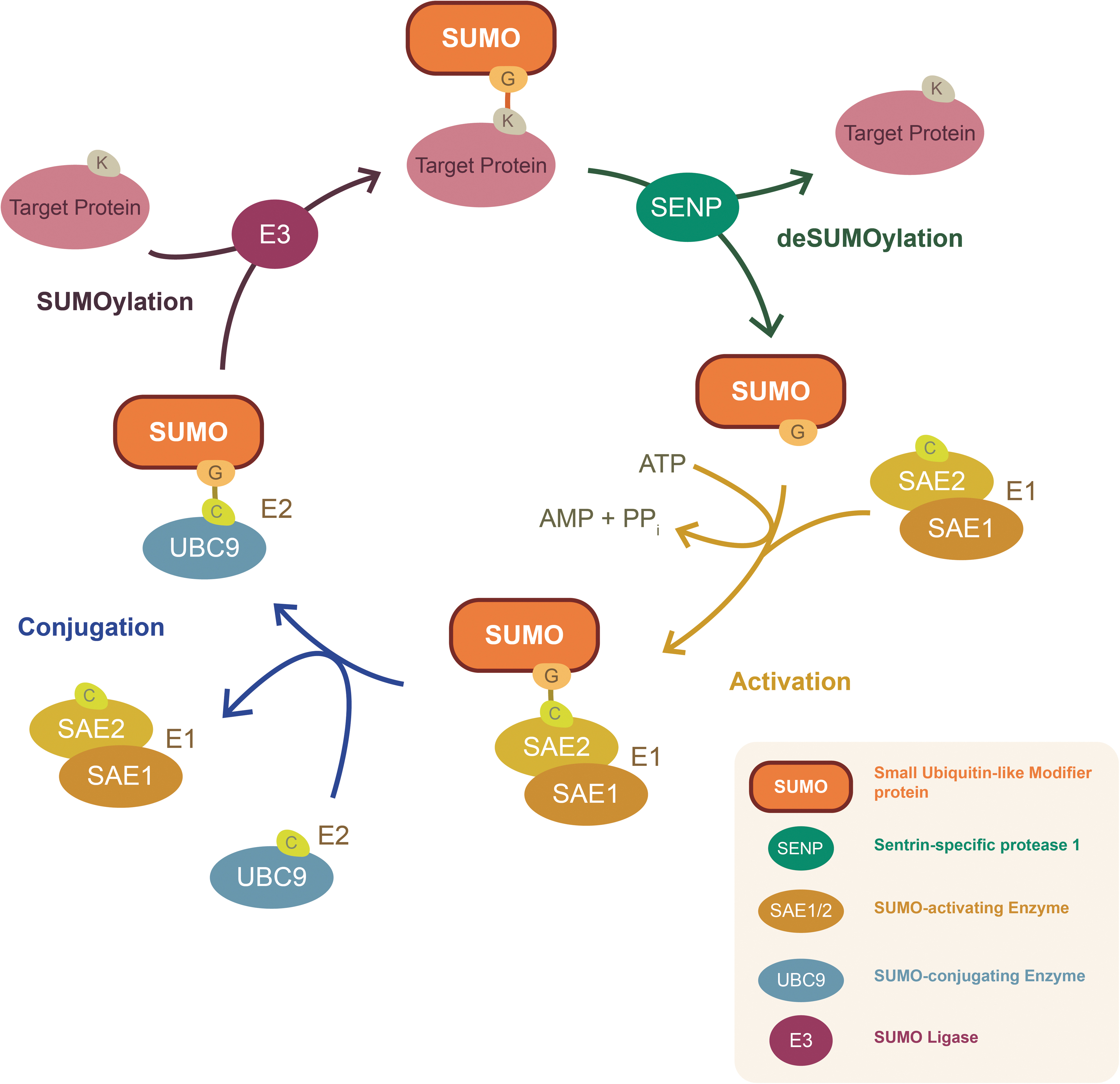
SUMOlyation cascade. Starting from the top right: precursor SUMO is processed to mature SUMO by multiple steps (1) cleavage by SENP, activation by E1 (SAE1/SAE2)+ATP, (2) transfer of SUMO to E2 (UBC9), and (3) ligation to target protein at a lysine (k) by E3 in the SUMOylation step (Top). Eventually SUMO is removed from the target protein by SENP in a deSUMOlyation reaction. E1, E1 activating enzyme; E2, E2 conjugating enzyme; E3, E3 ligase; SAE1, SUMO-Activating-Enzyme-Subunit-1; SAE2, SUMO-Activating-Enzyme-Subunit-2; SENP, sentrin-specific protease; SUMO, small ubiquitin-like modifier; UBC9, SUMO-conjugating enzyme UBC9.
SUMOylation Modulates Aging in C. elegans
Initially, Princz, et al. found that levels of SUMOylated proteins measured by Western blot peaked at day 4 of adulthood in C. elegans and then tailed off. This correlation with aging is clearly not complete as worms live only 12–18 days, the first clue that SUMOlyation might have a complex relationship with mammalian aging. These findings were followed up by studies of SUMO gene (smo-1) expression demonstrating a more consistent increase with age. Although complete knockout of smo-1 is embryonic lethal, RNAi knockdown of smo-1 reduced worm lifespan by ∼10%. By contrast, smo-1 overexpression extended lifespan by ∼10%. 4 Specific tissues are known to control worm longevity. 5 When knockdown of smo-1 was carried out in neurons, hypodermis, muscle, and intestine, only intestine and neuron-specific knockdown shortened worm lifespan. Interestingly, worm lifespan was only extended by smo-1 overexpression in the intestine, suggesting that intestine is the key target for lifespan extension.
Targets of SUMOylation to Benefit Lifespan
Heat shock factor 1 (HSF1), skinhead-1 (SKN-1 homolog of mammalian NRF2), and Abnormal DAuer Formation (DAF-16, homolog of FOXO-forkhead box proteins) have been identified as critical stress response (e.g., heat, oxidative challenge, and metabolic—insulin/IGF1) transcription factors that promote longevity. Prior studies demonstrated that HSF1 is SUMO modified in both C. elegans and mammals. 6,7 RNAi attenuation of SKN-1 expression shortens life and this effect is blocked by overexpression of SUMO, suggesting that SKN-1 contributes to the pro-longevity effects of SUMO, although perhaps SUMO could act downstream of SKN-1. DAF-16 is upregulated in C. elegans worms having loss-of-function mutations in the insulin/IGF1 receptor DAF-2. Smo-1 knockdown shortens the lifespan of DAF-2–deficient worms. Additional studies indicated that DAF-16, but not SKN-1, is directly SUMOylated. Moreover, Princz et al. observed that although SUMOlyation of DAF16 increased its nuclear localization, this actually decreased its transcriptional activity.
Mitophagy, a Specialized form of Autophagy
Mitochondria comprise tubular networks that are constantly undergoing fission and fusion. Dysfunction of mitochondria is a major hallmark of aging. 8 As the site of oxidative phosphorylation, mitochondria are particularly prone to oxidative stress. Aging disrupts the integrity of mitochondria contributing to the pathogenesis of many age-related disorders. 9,10 For example, mice with a defective proof-reading mitochondrial DNA (mtDNA) polymerase gamma exhibit an accelerated aging phenotype secondary to accumulation of mtDNA mutations. 11 Numerous studies correlate mtDNA deletions/mutations with aging, 12 central nervous system disease, 13 as well as lifespan and health span. 14 –16 The maintenance of healthy mitochondria contributes to successful cellular homeostasis and health span.
Autophagy, literally “self-eating,” was first described by Christian de Duve, 17 who won the 1974 Nobel prize for the discovery of lysosomes. Degradation of entire mitochondria by autophagosomes was observed by electron microscopy and thought to be a nonspecific process until Kissová et al. found that yeast mitochondrial protein Uth1p mediated selective mitochondrial degradation. 18 Although internal mitochondrial protein quality control and proteostasis are maintained by organelle-specific chaperones and the unfolded protein stress response (UPRmt), 19 the selective degradation of whole dysfunctional mitochondria by a process since termed “mitophagy” maintains the pool of healthy mitochondria. Inherited diseases with defects in mitophagy are characterized by complex multisystem pathologies, especially progressive neurodegeneration.
SUMOylation Regulates Mitophagy
Princz et al. found that levels of SUMOylation also increased within mitochondria during aging (Fig. 2). The dye, TMRE (tetramethylrhodamine, ethyl ester), stains mitochondria based on the mitochondrial membrane potential, ψ. ψ has been observed to decrease during aging of C. elegans. Knockdown of smo-1 alleviates the age-associated decline in membrane potential and smo-1 overexpression increases TMRE staining among young adult worms. 4 When both knockdown and increased expression of a gene affect rescue of a phenotype in the same direction, in this case rescuing decreased ψ, complex regulation is probably occurring. Because both DAF-16 and SKN-1 are known to be key regulators of mitochondrial homeostasis 20,21 and are regulated themselves by SUMOylation, studies were undertaken to directly connect smo-1 expression to the mitochondrial functions influenced by these genes. For this purpose, production of mitochondrial reactive oxygen species (ROS) can be measured by the MitoTracker ROS dye. Overexpression of smo-1 increased ROS production during aging. Inhibition of ROS by N-acetyl cysteine (NAC) treatment reduced the life extension observed among smo-1 overexpressors. This is a curious result to some extent as NAC has been reported to extend lifespan in C. elegans in other study. 22 Perhaps the benefit of NAC and smo-1 overexpression cancel each other out. Other studies of mitochondrial functions (e.g., ATP production and oxygen consumption) indicated that SUMO represses mitochondrial activity during aging through DAF-16.

Mitophagy pathways, There are two primary mitophagy pathways (Left). Ubiquitin-independent receptor-mediated mitophagy in the presence of normal/high membrane potential (ψ). In ubiquitin-independent receptor-mediated mitophagy, autophagy receptor proteins such as FUNDC1, BNIP3, or NIX are recruited to the membranes of the mitochondria. Finally, mitochondria are engulfed by autophagosomes (Middle). Upon mitochondrial damage with associated reduced membrane potential (ψ), ubiquitin-dependent PINK1/Parkin-mediated mitophagy is induced. At the outer mitochondrial membrane, PINK1 becomes stabilized, Parkin is activated, and mitochondrial proteins are ubiquitinated. Then, autophagy regulators p62, OPTN, and NDP52 together with LC3 help engulf mitochondria at the autophagosome. To form autophagosomes, the ER contributes autophagy core complexes including VPS34 and WIPI2, which form membranes and further recruit the ATG16L1 complex and LC3. At the terminal step, acidic lysosomes fuse with autophagosomes. ER, endoplasmic reticulum.
As animals age, the mitochondrial tubular meshwork becomes fragmented. 21,23 Such changes are alleviated in the SMO-1–depleted animals, indicating a requirement for SUMO in mitochondrial fission. RNAi knockdown of daf-16 augmented mitochondrial tubular network fragmentation in Smo-1–deficient animals during aging. DAF-16 controls a gene eat-3 (eating; abnormal pharyngeal pumping), a homolog of OPA1, a protein required for the fusion of the inner mitochondrial membrane. 24,25 EAT-3–deficient worms exhibit fragmented fragile intestinal mitochondria. Furthermore, RNAi inhibition of eat-3 in SMO-1–deficient animals triggers mitochondrial network fragmentation during aging.
Mitochondrial fission is required for initiation of mitophagy 26 (Fig. 2). Princz et al. 4 utilized the methods of Palakaris et al. 21 to monitor mitophagy. Knockdown of smo-1 inhibited mitophagy, which is consistent with the findings that reduced smo-1 blocks mitochondrial fission. Princz, et al. concluded that SUMO is required for mitochondrial fission in muscle and mediates its effects through DAF-16. 4 However, in the nervous system, RNAi knockdown of smo-1 had the opposite effect, it induced mitophagy, suggesting a more complex tissue-specific relationship 4 of SUMOlyation with mitophagy.
Princz et al. hypothesize that because animals with reduced SUMOlyation exhibit elongated mitochondrial morphologies with increased ROS, they are defective in maintaining mitochondrial homeostasis compared with mutants that maintain elongated mitochondria with reduced ROS and increased fusion as reported by Chaudhari and Kipreos. 24 It would be quite useful to carefully explore the differences between mitochondrial fission and fusion in smo knockdowns and overpressing animals to better understand how mitochondrial homeostasis is affected by SUMOlyation.
Medical Implications
Princz et al. provide data showing that levels of SUMO (smo-1) are increased during aging in C. elegans (although not the total amount of SUMOlayted proteins, which peak in middle age), somewhat similar to data in mouse plasma in which SUMOlyation increases into middle age and then somewhat decreases. 27 Princz et al. hypothesize that SUMOylation tunes the activity of “stress modulator proteins” (e.g., DAF16 and SKN-1) during aging to regulate cellular stress, augment mitophagy, and ultimately contribute to determining lifespan in worms. 4 As high as 15% of the human proteome is SUMOylated, making it quite likely many other targets will be identified. That so many proteins may be targeted suggests the hypothesis that loss of SUMOlyation or increased SUMOlyation due to overexpression is likely to affect processes that result in altered lifespan both positively and negatively, with the sum due to overexpression of smo-1 being net positive for lifespan increase in C. elegans. However, there is no guarantee that an analogous summation in mammals in response to increasing SUMOlyation would yield the same benefits.
The interplay between DAF16 activation versus SKN-1 activation is complex. Although DAF16 can activate SKN-1 and both cooperate to increase lifespan in daf-2 mutant C. elegans, and SKN-1 mammalian homolog, Nrf2 can rescue some pathologies associated with Hutchinson–Gilford progeria, 28 chronic constitutive activation of SKN-1 can deplete fat and even shorten lifespan in C. elegans. 29 In fact, although activation of SKN-1 is typically protective of various stress, long-term activation of SKN-1 actually reduces resistance to endoplasmic reticulum, mitochondrial and heat stress, while apparently suppressing DAF16 activity. 30 Thus, SKN-1 activation alone may be problematic. Only when it is combined with DAF16 activation, as observed in daf2 knockouts, are the negative consequences of SKN-1 on lifespan eliminated, and the combined action of SKN-1 and DAF16 capable of increasing lifespan. 30 Not only that, but also protection from oxidative stress through SKN-1 activation is not necessary for life extension by DAF16 overepression, 31 suggesting that protection from oxidative stress is not important to lifespan extension. Given these complex interactions between DAF16 and SKN-1, no wonder the role of SUMOlyation of these regulators is difficult to completely untangle.
Moreover, it is important to note that the effects of SUMOylation overexpression on lifespan in C. elegans are modest and pale in comparison with lifespan increases observed with the daf2 mutations, for example. Perhaps this is not surprising given that the direct SUMOlyation target is DAF16 and DAF16 overexpression itself only has a modest effect on lifespan at best. 32,33
In mammals, SUMOylation has been reported to regulate mitochondrial biogenesis through Drp1 and PGC-1alpha, 34 –36 suggesting some conservation of mechanism between invertebrates and mammals. However, no connection to lifespan enhancement has yet been reported and it is unclear what effect inhibition or activation of SUMOylation would have. The situation is even more complex in vertebrates, with control of these pathways having become more complex with specialization and redundancy in the IIS/FOXO family as well as the SUMO family. A role for SUMOlyation in vertebrate/mammalian cell phenotypes such as cell senescence has been described and effects on these more complex cell phenotypes are expected to occur given the large number of SUMOlyation targets. For example, SUMOylation of P53 and deSUMOylation of polycomb repressive complex member BMi1 regulate cellular senescence. 37
Inhibitors targeting protein SUMOylation have been of great interest to the pharmaceutical industry seeking novel cancer drugs 38 because of their reported anticancer activities. Moreover, in combination with Farnesoid X receptor agonists, SUMOylation inhibitors have activity against liver fibrosis in rodent disease models. 39 Normal patterns of SUMOlyation maintain neural function in mammals and evidence is accumulating that abnormal SUMOlaytion can promote neurodegeneration. 40 Interestingly the incidence of cancer, fibrosis, and neurodegeneration is associated with increasing age in humans.
Metabolic reprogramming of SUMOlyation for lifespan enhancement likely has intrinsic limitations and dangers in manipulation of these critical pathways. 41 Lesson for the interested: merely combining treatments, such as the NAC with smo-1 overexpression in C. elegans to attempt to extend lifespan may lead to disappointing results!
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.