
Abstract
Metformin, a commonly used well-tolerated treatment for type 2 diabetes, is being deployed in clinical trials to ameliorate aging in older nondiabetic humans. Concerningly, some experiments in model organisms have suggested that metformin use at old ages shortens life span and is toxic to mitochondria. The demonstrated safety of metformin therapy in humans and the conflicting data from model organisms compelled us to test the hypothesis that metformin treatment would be toxic to older rats. To define an effective dose in 30-month-old hybrid rats, we evaluated two doses of metformin (0.1%, 0.75% of the diet) and treated the rats for 4 months. Body mass decreased at the 0.75% dose. Neither dose affected mortality between 30 and 34 months of age. We assessed mitochondrial integrity by measuring mitochondrial DNA (mtDNA) copy number and deletion mutation frequency, and mitochondrial respiration in skeletal muscle and the heart. In skeletal muscle, we observed no effect of metformin on quadriceps mass, mtDNA copy number, or deletion frequency. In the heart, metformin-treated rats had higher mtDNA copy number, lower cardiac mass, with no change in mtDNA deletion frequency. Metformin treatment resulted in lower mitochondrial complex I-dependent respiration in the heart. We found that, in old rats, metformin did not compromise mtDNA integrity, did not affect mortality, and may have cardiac benefits. These data provide some reassurance that a metformin intervention in aged mammals is not toxic at appropriate doses.
Introduction
The geroscience hypothesis 1 considers age as the most significant contributor to many of the chronic diseases that exponentially accelerate human mortality. In accordance with this hypothesis, clinical trials are being implemented to modify the rate of aging and reduce the burden of chronic disease. 2 Proposed medications include metformin, rapamycin, and resveratrol, all molecules that target metabolism. 3 –6 Metformin treatment in older adults is being tested in 65–79-year-old nondiabetic adults to improve human health span. 7
As these treatments are being deployed in older adults who are otherwise healthy, it is important to have a clear understanding of the side effects and associated risks of treatment. Metformin is a widely prescribed essential medicine for the treatment of diabetes mellitus type 2 (DM2) with an extensive safety record. Despite its widespread use, the molecular mechanisms of metformin are not yet fully understood.
Metformin has pleiotropic effects on metabolism including activation of AMP kinase, inhibition of the mitochondrial respiratory chain complex I, and glucagon signaling through cAMP. 7 Aging also affects metabolism through innumerable molecular, cellular, and tissue-level alterations that have not been fully elucidated. 8 Better understanding of how metformin intersects with the processes of aging would clarify its safety profile for nondiabetic adults.
In preclinical longevity studies in invertebrates and rodents, the potential benefits and risks of metformin treatment have had mixed effects. In nematodes, lifelong metformin treatment increased life span. 9,10 Lifelong treatment of mice with metformin extended life span in two trials, 11,12 but no effect was observed in a third study. 13 In contrast to the lifelong treatment of mice, initiation of metformin therapy for human aging is being proposed at older ages, which is being tested in animals. 7 In mice, metformin treatment initiated at older ages did not extend life span 12,14 and, in one study, increased mortality. 15 In Caenorhabditis elegans, metformin treatment initiated at old ages was toxic. 10 The life span effects of metformin remain unclear and may depend on the age at which treatment is started.
The risks and benefits of metformin treatment have been species-, dose-, route-, and age-dependent. In mice, a 1% metformin dose in the diet was found to be toxic, whereas 0.1% was well tolerated and extended life span. 11 By contrast, a 1% dose was better tolerated if administered episodically to 24-month-old mice. 14 In 3-month-old Zucker rats, metformin impaired mitochondrial respiratory chain activity at 100 and 300 mg/kg, but not at 30 mg/kg. 16 In 3-month-old Sprague-Dawley rats, treated for 3 months, the no-observed-adverse-effect level for rats was 200 mg/kg. 17
Late-life metformin treatment of C. elegans and aged female mice induced deleterious changes in conserved metabolic pathways and showed defects in electron transport and ATP production and cardiomyocyte toxicity. 10,15 A contributing factor to the age-dependent effects of metformin may be the concurrent age-induced decreases in mitochondrial integrity, 18 which increases the susceptibility of older adults to the adverse effects of treatments that affect mitochondria. Mitochondrial integrity has not been examined in aged mammals after metformin treatment. We hypothesized that metformin initiated at old ages would compromise mitochondrial integrity and that this would be reflected in indices of mitochondrial DNA (mtDNA) quantity and quality, and mitochondrial function.
We examined metformin's effects on mitochondrial integrity when given at 30 months of age to a well-characterized model of mammalian aging, the Fischer 344 x Brown Norway F1 (F344xBN F1) hybrid rat. 19 Then 30-month-old rats are comparable with ∼65-year-old humans, based on survival. We chose to treat the rats orally with two doses of metformin. The first dose of 0.1% has been used in mice, is well tolerated, and extended life span in some studies. 11 A second dose of 0.75% was chosen as it approximates, in rats, the dosage to be used in the proposed targeting aging with metformin (TAME) clinical trial—1700 mg/day. 7 Indices of mitochondrial integrity included mtDNA copy number, mutation frequency, and mitochondrial respiratory function, all of which are perturbed by age.
mtDNA copy number declines with age are controversial, however, we and others have consistently observed decreases in skeletal muscle mtDNA copy number with age. 20 –22 Concomitantly, mitochondrial respiratory chain activity decreases with age. 18 In contrast to these decreases in mtDNA copy number and enzymatic activity, mtDNA deletion mutations increase exponentially with age in muscle. 21 If metformin has toxic effects on mammalian mitochondria, then it should exacerbate age-sensitive mitochondrial defects that are reflected in the aforementioned metrics.
Here we show that 0.75% metformin treatment initiated in aged rats recapitulates known effects on body mass and mitochondrial respiratory chain complex I-dependent respiration without negative effects on survival, skeletal muscle mass, mtDNA copy number, or mtDNA deletion mutation frequency. Positive effects of metformin treatment in aged male rats included reduced cardiac mass and increased heart mtDNA copy number, which further supports the use of metformin for age-induced cardiovascular disease. 23 Our data show that an appropriate dose of metformin does not compromise mitochondrial integrity in old male rats and may benefit older individuals without diabetes.
Materials and Methods
Animals, metformin treatments, and tissue preparation
This study was carried out in accordance with the recommendations in the National Institutes of Health (NIH) Guide for Care and Use of Laboratory Animals and the guidelines of the Canadian Council on Animal Care. The protocols used were approved by the Institutional Animal Care and Use Committees at the University of Alberta and UCLA. Thirty-month-old male F344xBN F1 hybrid rats were obtained from the NIA Aging Rodent Colony. Twenty-three rats were fed a control diet, 12 rats were fed metformin 0.1% in the diet, and 13 rats were fed 0.75% in the diet for 4 months.
Metformin was purchased from Spectrum Chemical (New Brunswick, NJ). Metformin containing chow was formulated to 0.1% or 0.75% by weight in NIH-31–irradiated diet (Envigo, Madison, WI). Rats were housed on a 12-hour light/dark cycle. Animals were euthanized by carbon dioxide asphyxiation followed by exsanguination. Tissues were dissected from the animals, weighed, and flash frozen in liquid nitrogen, and stored at −80°C.
DNA isolation
Rat quadriceps muscle or heart was ground to a powder using a mortar, pestle, and liquid nitrogen. Total DNA was extracted using proteinase K digestion with sodium dodecyl sulfate and ethylene diamine tetraacetic acid, phenol/chloroform extraction, and ethanol precipitation as previously described. 20 Total DNA quantity and quality were measured using spectrophotometry at A230, A260, and A280 (ThermoScientific Nanodrop 2000 Spectrophotometer), fluorometry (ThermoFisher Qubit 2.0 Fluorometer), and integrity examined by gel electrophoresis or TapeStation 4200 (Agilent).
mtDNA copy number and mtDNA deletion frequency by digital polymerase chain reaction
A 5-prime nuclease cleavage assay and droplet-based digital polymerase chain reaction (dPCR) were used to quantitate copy numbers for nuclear DNA, total mtDNA, and mtDNA deletions with specific primer/probe sets for each as previously described. 20 mtDNA deletion frequency is the proportion of mutant molecules per wild-type mtDNA. Deletions per 100 nuclei value is normalized to the single copy nuclear gene, Hprt1. dPCR quantitation of all samples and for all targets was performed on blinded samples.
Respirometry assay on frozen tissue samples
Respirometry was performed on frozen skeletal muscle and cardiac tissue according to a validated protocol. 24 In brief, frozen tissues were thawed in ice-cold phosphate-buffered saline, minced, and homogenized. Protein concentration was determined by bicinchoninic acid assay (ThermoFisher). Mitochondrial respiratory chain oxygen consumption rate (OCR) through complexes I, II, and IV was measured using a Seahorse XF96. Seahorse Wave software (Version 2.6.1, Agilent) was used to export OCRs normalized by protein concentration.
Statistical analysis
All data are presented as means ± standard error of mean. Data were tested for a normal distribution. Student's t-test was used to compare differences between treatment and control groups. Non-normally data were analyzed by Mann–Whitney tests. Log-rank test was used to analyze survival curves. Prism (Version 7.05, GraphPad Software) was used to compute p-values.
Results
Morphometric measures after metformin treatment
Ninety percent of F344xBN F1 hybrid rats survive to 29 months of age and 34 months of age is the median life span. Thus, during the treatment period, from 30 to 34 months of age (corresponding to ∼65–78 years of age in humans), the rats are vulnerable to age-induced morbidity and mortality. Despite this, no difference was observed in the survival curves for either metformin dose (0.1%, 0.75%) compared with control (log-rank, p = 0.5808). Food consumption was not affected by the 0.1% metformin dose. Food consumption was, on average, 26.5% lower for the first 7 weeks in the 0.75% metformin treatment group, but did not differ by the end of the 4 months of treatment.
The decreased food consumption in the 0.75% metformin group mirrored the body mass loss. Body mass changes after metformin treatment are well-described benefits in adults with DM2. 25 –29 Modest weight loss suggests an efficacious dose in rodents. In contrast to body mass, muscle mass loss has not been observed with metformin therapy in older adults with DM2. 26 After 4 months of treatment in aged rats, metformin had dose- and tissue-dependent effects on body, quadriceps, and heart mass (Fig. 1). All rats lost body mass between 30 and 34 months of age. On average, rats treated with 0.75% metformin lost 23.5% of their body weight (from 535 ± 30 g to 409 ± 36 g), whereas controls lost 10.6% (from 537 ± 38 g to 480 ± 38 g).
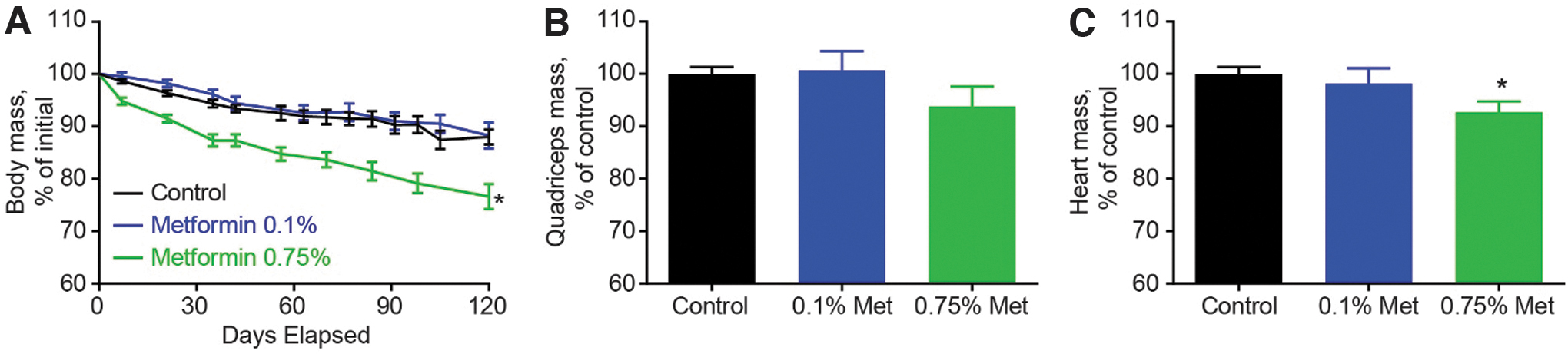
Morphometric measures after metformin treatment at two doses.
There was no difference in body mass loss between controls and 0.1% metformin-treated rats (from 511 ± 53 g to 450 ± 49 g). There was no effect of metformin on quadriceps mass at either dose (p = 0.863 for the 0.1% dose; p = 0.160 for the 0.75% dose). At 34 months, the heart mass was greater by 7.8% (p = 0.032) and 1.8% (p = 0.601) in the control rats as compared with those fed 0.75% or 0.1% metformin, respectively. Based upon these data, we further examined the effect of 0.75% metformin on mtDNA integrity and mitochondrial function.
mtDNA integrity in quadriceps muscle and heart after metformin treatment
Four months of metformin treatment in 30-month-old rats had no effect on quadriceps mtDNA copy number (p = 0.381), deletion mutation frequency (p = 0.296), or deletions per 100 nuclei (p = 0.179) (Fig. 2). In the heart, 4 months of metformin treatment at old age did not alter mtDNA deletion mutation frequency (p = 0.618) or deletion mutations per 100 nuclei (p = 0.416) (Fig. 2). In contrast to skeletal muscle, heart mtDNA copy number was 17% higher in the 0.75% metformin-treated rats than in the control rats (p = 0.0376). In all samples, mtDNA deletion mutations per 100 nuclei were higher in the quadriceps than in the heart.
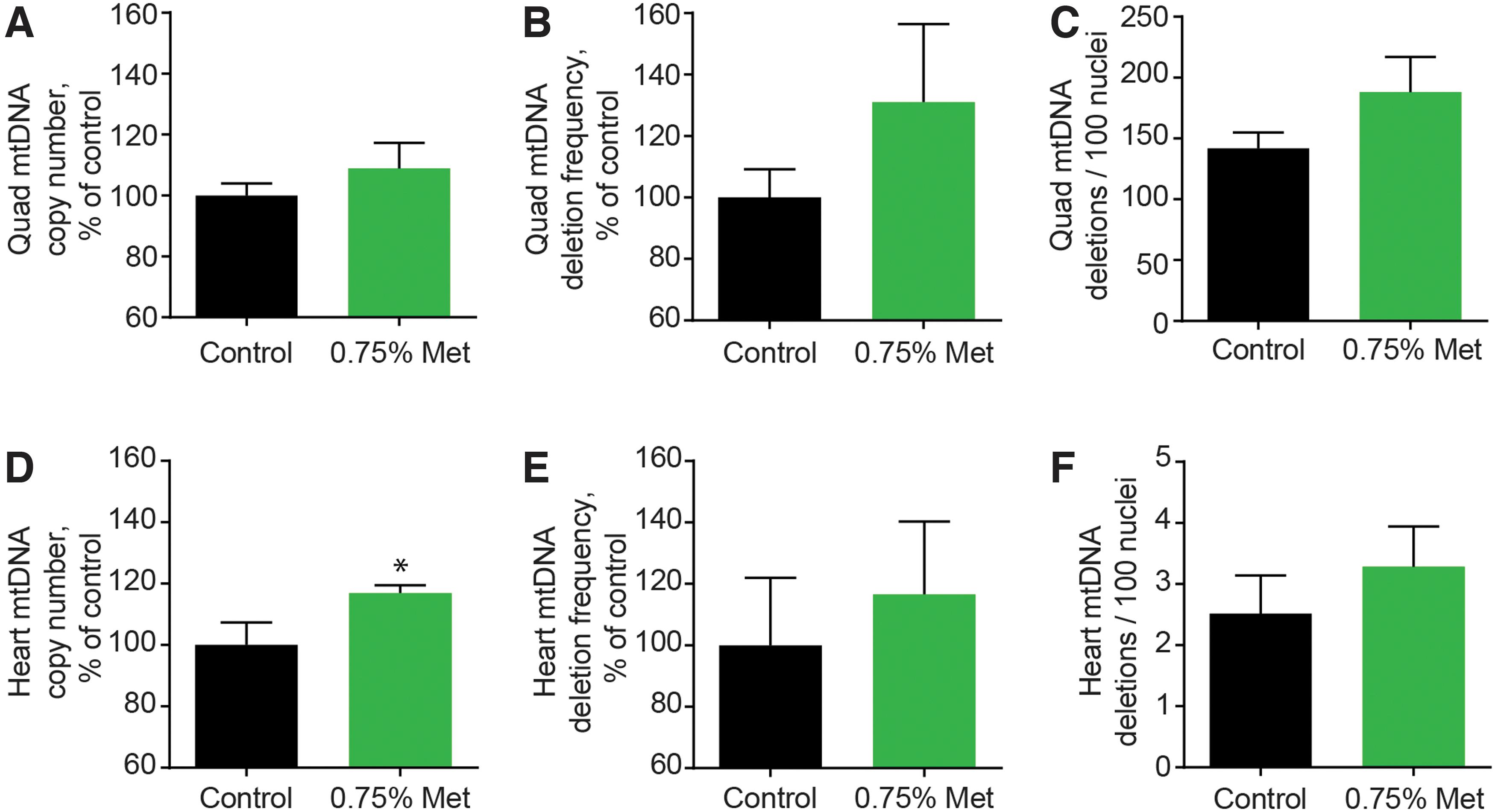
mtDNA quantity and quality after metformin treatment in quadriceps muscle and heart.
Mitochondrial function and content after metformin treatment
Complex I-dependent respiration is decreased with both age and metformin treatment. 30 However, in 34-month-old male rats, quadriceps muscle mitochondrial respiration was not different after metformin treatment (complex I dependent [p = 0.151]; complex II dependent [p = 0.139]; and complex IV dependent [p = 0.133]). By contrast, metformin treatment decreased heart complex I-dependent respiration by 28.4% (p = 0.0082) (Fig. 3A, C).
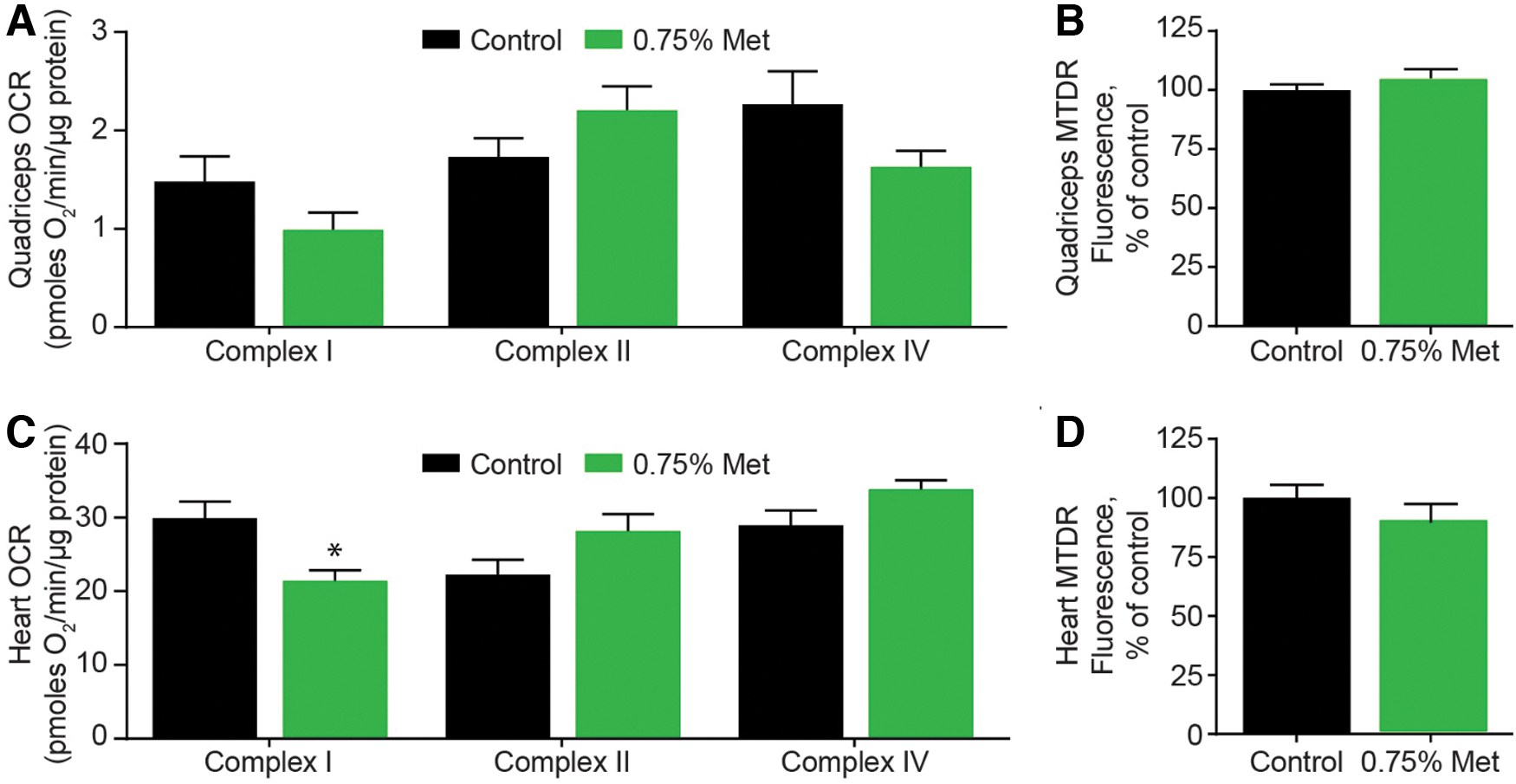
Mitochondrial function and content in the quadriceps and heart after metformin treatment in 34-month-old rats.
We assessed mitochondrial content by three different methods: (1) mtDNA copy number, (2) complex IV-dependent respiration, and (3) mitochondrial staining with MitoTracker Deep Red (MTDR). Complex IV-dependent respiration and mtDNA copy number were 12.8-fold and 21.6% higher, respectively, in the control heart versus control quadriceps. By contrast, mitochondrial staining intensity with MTDR was not different between the control heart and quadriceps (data not shown) or between metformin-treated and control rats at 34 months of age in quadriceps (p = 0.281) and heart (p = 0.283) (Fig. 3B, D). Metformin had no significant effect on these measures of mitochondrial content in 34-month-old rats.
Discussion
We hypothesized that metformin started at old ages would compromise mitochondrial integrity based on previously reported data. 10,15 Our data show that an appropriate dose of metformin (as discussed hereunder), initiated at old age, does not compromise mitochondrial integrity and may have cardiac benefits for older nondiabetic individuals. In addition, we did not observe increased morbidity or mortality in the metformin-treatment groups.
In another study, 1% metformin treatment initiated at 24 months of age in mice showed comparable life spans between metformin and control groups. 14 In the late-life metformin study in C. elegans, life span was used as the index for metformin dose. 10 This approach is not feasible in human trials. Our indicator for drug efficacy was body weight loss, which also occurs in humans. The weight loss was accompanied by reductions in food consumption as has been observed in other rat strains 31,32 and in humans 33 –35 with metformin treatment.
Short-term caloric restriction has cardiac benefits in rats, but the degree of caloric restriction has been greater than what occurred in this study. 36,37 This two-dose study in hybrid rats was not powered to detect increased survival over the 4-month treatment and observation period. Measuring the dose-dependent health span and life span effects of metformin treatment in rats may help clarify the benefits and risks of metformin therapy for aging humans.
As a wide variety of doses have been attempted in numerous species to affect aging and disease, we sought to establish an appropriate dose for aged rats. Our criteria were (1) to avoid metformin-induced mortality in our experimental animals, as this was a noted concern in mice at a dose of 1% metformin in the diet 11 and (2) that metformin should induce body mass loss as this is an effect of metformin therapy in humans with and without diabetes. 29,34 When adapting dosages between mammals, adjustments are typically made for the relative surface area-to-weight ratio. 38
The ratio of surface area-to-weight ratio adaptation is a reflection of the metabolic rate differences between mammals that influences pharmacokinetics and is critical for determining a therapeutic dose. 39 Smaller mammals have lower surface area-to-volume ratios and higher metabolic rates. Metformin targets metabolism and thus the therapeutic effects, and toxicities, of metformin will depend on the species- and age-specific metabolic rate. For example, mice require high metabolic rates to maintain body temperature and thus metformin may have higher toxicity in mice as compared to rats or humans, which have lower basal metabolic rates.
In mice, a dose of 0.1% (formulated in chow) reduced body mass, 11 but we observed no such effect in rats. The 0.75% therapeutic dose chosen for this study approximates the dose proposed for the TAME trial on a surface area-to-weight ratio, but the higher metabolic rate of rats suggests that the therapeutic window in rats is narrower than in humans. Body mass in obese humans decreased by ∼5% with metformin treatment, 29 whereas in our study, at comparable doses, rat body mass decreased 23.5%. Therefore, the rats in this study were exposed to higher effective doses than proposed for humans in the TAME trial. 7 Metformin's dose needs to be carefully titrated to match the underlying metabolic rate of each species and age. It is less clear how a therapeutic dose would be extrapolated to ectotherms.
In young nematodes, metformin increases life span in a dose–responsive manner. 9,10 When these same doses were used in old worms, mitochondria were impaired, resulting in toxicity. 10 If metformin had mitochondrial toxicities in older mammals, one would expect pronounced effects in heart and skeletal muscle as they depend on oxidative phosphorylation, have high mitochondrial content, and exhibit age-induced mitochondrial impairments. 18 mtDNA integrity was not decreased in skeletal muscle or heart after metformin treatment in old rats. We did, however, observe a statistically significant decrease in complex I-dependent respiration in the heart, an expected effect of metformin treatment that indicates an effective dose was used in this study.
Metformin has mixed effects on aging skeletal muscle. In contrast to life-long treatment, late-life initiation of metformin treatment in mice altered mitochondrial gene expression. 14 In healthy older humans, metformin blunts the hypertrophic response of skeletal muscle to resistance training, 40 but reverses the gene expression alterations of aging. 41 Despite these findings, no negative impact on lean mass was observed after 18 months of metformin treatment in human subjects with an average age of 60 years. 26 We did not observe metformin-induced alterations on rat quadriceps muscle mass, mitochondrial integrity, or function. Overall, metformin appears to have modest mitochondrial effects at old age in skeletal muscle.
The cardiac effects of metformin treatment in old rats may have clinical implications for patients with heart failure. Decreased ATP synthesis has been observed in mouse models of cardiac hypertrophy 42 and human subjects with heart failure with preserved ejection fraction have reduced cardiomyocyte energy reserves. 43 Moreover, in a mouse model of heart failure, treatment with metformin improved left ventricular function, mitochondrial respiration, and ATP synthesis. 44 As in humans, cardiac hypertrophy is a well-described phenotype of aging in the F344xBN F1 hybrid rats. 45 In contrast to skeletal muscle and body weight, cardiac mass increases with age. This increased cardiac mass was blunted by metformin treatment and suggests beneficial cardiovascular effects 46,47 and a possible positive clinical outcome from metformin in humans. 48
Conclusion
Metformin has been proposed as an intervention to ameliorate the metabolic and cardiovascular derangements associated with aging. 49 An incomplete understanding of the molecular targets of metformin and how these intersect with aging, coupled with unclear preclinical data on lifespan benefits and/or toxicity is complicating the use of metformin as a geroprotector in older adults. In our study, we observed no overt mitochondrial toxicity in skeletal muscle or heart after metformin treatment at old age in rats. We observed modest effects in the heart that could be interpreted as beneficial.
Footnotes
Authors' Contributions
Study design and conception were carried out by A.H. and J.W. Experiments were performed by C.K., A.H., and A.H. Data analysis was done by by A.H. and J.W. All authors contributed to writing and revising the article.
Ethical Approval
This study was carried out in accordance with the recommendations in the NIH Guide for Care and Use of Laboratory Animals and the guidelines of the Canadian Council on Animal Care. The protocols used were approved by the Institutional Animal Care and Use Committees at the University of Alberta and UCLA.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study is supported by the National Institute on Aging at the National Institutes of Health (Grant Nos. R56AG060880, R01AG055518, K02AG059847, and R21AR072950).