
Abstract
Epigenetic alterations during aging are manifested with altered gene expression linking it to lifespan regulation, genetic instability, and diseases. Diet and epigenetic modifiers exert a profound effect on the lifespan of an organism by modulating the epigenetic marks. However, our understanding of the multifactorial nature of the epigenetic process during aging and the onset of disease conditions as well as its reversal by epidrugs, diet, or environmental factors is still mystifying. This review covers the key findings in epigenetics related to aging and age-related diseases. Furthermore, it holds a discussion about the epigenetic clocks and their implications in various age-related disease conditions, including cancer. Although, epigenetics is a reversible process, how fast the epigenetic alterations can revert to normal is an intriguing question. Therefore, this article touches on the possibility of utilizing nutrition and mesenchymal stem cell secretome to accelerate the epigenetic reversal and emphasizes the identification of new therapeutic epigenetic modifiers to counter epigenetic alteration during aging.
Introduction
Aging is a progressive decline in the physiological process, which leads to the onset of various diseases and disorders such as diabetes, cancer, and cardiovascular diseases and the risk of higher mortality rate. The probability of cancer increases after 50 years of age while heart diseases increase after 60 years of age. Thus, aging has the foremost impact on the social/economic welfare of an individual. 1 Hence, there is a dire need for rejuvenating factors that can delay aging and extend lifespan. In the last few decades, life expectancy has increased widely due to the significant improvement in medical health facilities and health awareness among individuals. 2 However, the mechanism underlying aging is still unclear. Various studies are undergoing to delineate the cellular and molecular hallmarks of aging. Epigenetic modification is considered to be the vital mechanism that influences the gene expression associated with aging and age-related disorders such as heart failure and cancer. 3,4 Epigenetic factors influence gene regulation by regulating the binding of transcriptional factors to the DNA.
These epigenetic factors include enzymes such as methyltransferase and demethylase that modify the methylation status of the DNA or core histone subunits H2A, H2B, H3, and H4 as well their variants H3.3, macroH2A, and H2A.Z. Emerging evidence support that aging is associated with the gradual loss of histones, chromatin remodeling, DNA, and histone modifications. 5,6 Due to the reversible nature of epigenomics, the current focus is on delineating the epigenetic changes involved in aging to develop novel therapeutic targets to circumvent aging and age-related disorders. 5,6 This review covers the key finding in epigenetics related to aging, age-related diseases, and how epigenetic modification have an impact on lifespan and health span extension. It will further discuss epigenetic clocks and their implication in various age-related disease conditions, including cancer. Finally, it will shed light on the role of nutrition, including methionine, folate, caloric restriction, diet, and mesenchymal stem cell (MSC) exosomes as epigenetic modifiers in combating aging.
Epigenetics and Aging
Aging and lifespan have been linked to epigenetic modifications in many organisms. Various studies suggest that epigenetic modifications either lead to the alleviation of aging, delay age-associated disorders, and increase the lifespan of organisms or promote aging and decrease the lifespan of organisms. Therefore, understanding the mechanism underlying the epigenetic modifications in aging will provide new insight into the development of strategies that could delay aging and age-associated disorder and extend lifespan and health span.
Histone Modifications
Histone modification at the post-translational level includes methylation, acetylation, phosphorylation, and ubiquitination. Among these, histone modifications at lysine residue, including methylation and acetylation are widely studied and known to influence the aging process. 7
Influence of Histone Modifications on Aging and Lifespan of Different Species
Histone methylation such as H3K4me3, H3K36me3, and H3K79me3 are associated with transcriptional activation while H3K27me3, H3K9me3, H4K20me3 are involved in the transcriptional repression and these modifications can drive the aging process by regulating the genes related to the same. 8 A study showed that knockdown or inhibition of mRNA level of H3K4 mono- and di-demethylase LSD-1 increases the lifespan in C. elegans. 9 Also, it was seen in yeast that mutation in COMPASS (H3K4 methyltransferase complex) influences the expression of 500 genes and reduces the replicative lifespan. 10 It was found in S. cerevisiae and C. elegans that loss of H3K36me3 is associated with a shorter lifespan or deletion of K36me2/3 demethylase Rph1 in yeast is associated with increased expression of H3K36me3 and longer lifespan. 7 Knockdown of UTX-1 extends the lifespan of worms with an increase in the global expression of H3K27me3. 11 It was observed in drosophila that hypomethylation of H3K27me3 is associated with longevity and a healthy life span by promoting glycolysis. 12
A study stated global reduced expression of H3K27me3 in the prematurely aged cells of patients with Hutchinson–Guildford progeroid syndrome (HGPS). 7 Liu et al. observed increased expression of H3K27me3 in the quiescent satellite cells of mice. 13 Another study showed that loss of H3K9me3 in adult drosophila midgut was associated with intestinal stem cell aging. 7 It was observed that H3K9me3 methyltransferase SUV39H1 was downregulated in both human and mouse hematopoietic stem cells (HSCs) with age, which corresponded to global decreased expression of H3K9me3 and impaired heterochromatin function, which is a marker for aging. 14 It was found that the H3K4me3 level was upregulated in mouse HSCs with age. 15 However, H3K4me3, H3K4me1, and H3K27ac were found downregulated in human HSCs with age. 7 These studies indicate that mice and worms exhibit a different pattern of H3K27me3 during aging as compared with human and drosophila, while mouse and human HSCs showed distinct expressions of H3K4me3 during aging indicating that histone marks may play an opposite role in controlling aging across different species.
Acetylation of a histone lysine results in the neutralization of its positive charge, which weakens the interaction between histone and DNA and leads to transcription activation. 8 Histone acetyltransferases (HATs) are known as transcription activators while histone deacetylases (HDACs) are regarded as corepressors and both play role in aging and longevity. 8 It was found that aged mouse hepatocytes exhibit decreased expression of H3K9me3/H3K14ac bivalent marks suggesting that histone acetylation is associated with aging. 7 Sirtuins are class III HDAC that play a role in the regulation of deacetylation of a lysine residue in an NAD+-dependent manner. 7 Among sirtuins, SIRT1 was found to be downregulated in various tissues of mice and humans, including the heart, liver, kidney, and brain with age. 7 Sirt6 is an H3K9 deacetylase that was shown to inhibit senescence when overexpressed in rat and human nucleus pulposus cells (NPCs) and contributes to longevity. 16 It was found that the knockdown of H3K56ac, Hst3, and Hst4-related HDAC-encoding genes during yeast aging is associated with a shorter lifespan. 17
SIRT2 level is essential for the rejuvenation of oligodendrocyte progenitor cells and is associated with remyelination in aged mice. 18 These reports suggest that increased expression of HDACs is associated with a longer lifespan and can be used as therapeutic targets to boost the lifespan of an organism.
Histone Methylation in Age-Related Pathological Conditions
Aberrant histone methylation directly contributes to age-related pathological conditions by dysregulating several signaling pathways, which results in the development and progression of several pathological conditions, including cardiac disorders, neurological disorders, osteoporosis, and cancer. 19 Lyu et al. showed that loss of H4K20me3 is associated with cardiac aging and is regulated by transforming growth factor (TGF)-β signaling through miR29a. 19,20 Global H3K79 hypermethylation was observed in the neurons of aged individuals, which was inversely coupled to the energy metabolism suggesting its role in brain aging and age-related brain disorders. 21 Zhang et al. found a decrease in the expression of H3K27me3 levels and an increase in the expression of H3K4me3 in neuroinflammation-associated genes of 22-month-old rats.
Furthermore, they noted that H3K27me3 was downregulated in intracerebral hemorrhage (ICH)-induced acute brain injury of the young and old rats, whereas H3K9ac levels were increased in 22-month-old rats after ICH, whereas H3K4me3 levels were unchanged in the young and old rat after ICH suggesting H3K27me3 and H3K9ac as the possible epigenetic target for the treatment of age-related brain injury and neuroinflammation. 22 It was found that aged mice exhibited reduced expression of SUV39H1 histone methyltransferase (HMT) and upregulation of Mkp-1 in the hippocampus, which was associated with a depressive-like phenotype. 23 Alzheimer's disease (AD) is observed mainly in the elderly and is related to dementia. 24 Lee et al., observed elevated levels of H3K9me3 in the cortical neurons of AD patients, which were associated with abnormal heterochromatin structure and synaptic dysfunction. 24 These findings indicate distinct expression of H3K9me3 in the different regions of the brain, which is associated with different pathological conditions. Spinal stenosis is an age-related degenerative disorder commonly found in the elderly population. 25
It was found that H3K4me3, H3K36me3, H3K9ac, and H3K18ac were highly upregulated in the fibrocartilage area adjacent to the degenerated irregular ligament in the ossification of ligamentum flavum (OLF) rat model. 25 Furthermore, WNT5A and GDNF were found hypermethylated in non-OLF MSCs than OLF MSCs suggesting their role in the regulation of osteogenic genes, including RUNX and BMP2, in spinal ligament ossification. 25 Various studies showed that aberrant histone modification and DNA methylation of critical genes, including WNT5A, GDNF, ACSM5, miR-497 and miR-195, increase spinal ligament degeneration. 25
Histone Acetylation in Age-Related Pathological Condition
Altered histone acetylation has been associated with large-scale chromatin modifications and thus can influence gene expression. 26 These modifications have been observed in aging tissues and various age-related pathological conditions, such as neurological disorders, cardiac disorders, cancer, and liver disorder. 26 It was found that sirtuins genes, including sirt2, sirt3, sirt4, and sirt6, were differentially expressed in aged mice and were associated with loss of cochlear hair cells. Among these, sirt6 was found to be upregulated during aging and suggested as a potential biomarker and therapeutic target for aged-related hearing loss. 27 It was found that sirt2 inhibition was associated with the amelioration of cognitive function and Aβ pathology in the AD mouse model. 28 A study reported that cognitive impairment can be prevented in aged mice by activating the AMPK-SIRT1-PGC1α pathway and antagonizing oxidative stress. 29 A ketone ester-rich diet-mediated increased expression of sirt3 prevents degeneration of GABAergic neurons and seizure-related death in AD mice. 30
Song et al. reported that AMPK/Sirt1-mediated inflammation was increased in aging rats and was positively correlated with myocardial fibrosis. 31 A study found reduced vascular expression of sirt1 in old obese individuals implicated in microvascular dysfunction through controlling mitochondrial reactive oxygen species levels and proteins involved in the mitochondrial respiration chain. 32 Li et al., observed that sirt6 suppressed charged multivesicular body protein 2B (CHMP2B) in mouse aged hearts associated with aging-related intolerance to ischemia/reperfusion injury. 33 Zhu et al., observed that inhibiting HDAC3 in the hippocampus reduces spatial memory deficits and reduces amyloid plaque load and Aβ levels in the brains of AD mice model. 34
A study stated downregulation of acetylation of H3 histone at K9, K18, and K27 in human-induced pluripotent stem cell-derived hepatocyte (hiHep) aging. Furthermore, they showed that increasing the acetylation of histones using HDAC inhibitors (HDACi), including Nab and valproic acid (VPA) resulted in increased proliferation of aged hiHep (> = 40 days) suggesting epigenetic modifiers as probable therapeutic targets for ameliorating aging and age-associated disorders such as reduced hepatocyte plasticity. 35
These research reports suggest chromatin remodelers, including SIRT1 and SIRT6 as important therapeutic targets for longevity and better health management for preventing the deterioration of various cells, tissues, and organs of the body. Therefore, it would be interesting to delineate the role of histone modifications in DNA repair, metabolism, inflammation, and oxidative stress in combating aging. It would be worthwhile to explore the most prevalent histone marks among different age-related disorder associated with aging process and the epigenetic modifiers for the same.
Influence of DNA Methylation on Aging and Age-Related Disorders
DNA methylation is a chemical modification that occurs mostly on the 5-carbon of cytosine island (CpG dinucleotide) identifiable by microarrays, deep sequencing, bisulfite conversion coupled to PCR, LC-MS/MS, and pyrosequencing. 36 –39 DNA methylation/demethylation is associated with transcription repression/activation. DNA methylation is absent in yeast and appears limited in Drosophila melanogaster. 40 Instead of DNA cytosine methylation (5 mC), N6 adenine methylation (6 mA) has been observed in worms. 41
It is well known that transposable repetitive elements, including Alu and LINE-1, harbor more than 90% of genomics 5-methylcytosines within their CpG islands and have been used to estimate the DNA methylation of the genome. 42 Various studies have shown that both Alu and LINE 1 repetitive element exhibit decreased methylation and variability with age suggesting aging is associated with DNA hypomethylation. 43 Other studies found that DNA hypomethylation at specific regulatory and repetitive elements such as Alu elements (Alu) and long interspersed element-1 (LINE1) leads to genetic instability and subsequent tumor formation, which is an age-related disorder. 44,45
It was observed that IL-7 mediated a decrease in the expression of DNMT3a and DNMT3b and an increase in the expression of TET2 and TET3 led to the increase in the demethylation of Bcl-2 and c-Myc in aged mice, which resulted in the increased population of DN3 cells associated with thymic involution. 46 It was found that individuals over 100 years of age exhibited whole-genome DNA hypomethylation in CD4+ T cells as compared with newborns. 47 A decrease in 5 mC was also observed in the various organs, including the liver, brain, and small intestinal mucosa of old mice as compared with young mice. 48
It is well known that aging predisposes organisms toward various abnormalities, including cancer, type 2 diabetes, and dementia. A recent report predicted altered DNA methylation status in several dementia-related genes, including PON1, AP2A2, MAGI2, POT1, ITGAX, PACSIN1, SLC2A8, and EIF4E, in healthy older adults >70 years. Among these, some of them developed dementia in later life suggesting some of the age-associated abnormalities can be detected early and raises the possibility of their prevention with epigenetic modifiers. 49 Chinn et al. noted global DNA hypomethylation in aging mice and increased DNA methylation variability among male and female mice in the mouse dorsal hippocampus. 50 Hahn et al. observed that DNMT1 is associated with age-related loss of cortical inhibitory interneurons, which is associated with cognitive decline. 51 Huang et al. demonstrated that loss of DNA methylation activity due to proteasomal degradation of DNMT3a variants drives the expansion of hematopoietic stem cells implicating the increased risk of age-related hematologic disease through epigenetic alterations. 52
Several reports have identified DNA methylation alteration in various tissues of T2D patients, including blood, pancreas, skeletal muscle, liver, and adipose tissues indicating that epigenetic modification during aging can prone individuals toward the development of type 2 diabetes. 53 These investigations signifies that DNA hypomethylation is associated with aging and can lead to age-related abnormalities and therefore also raises the possibilities of their prevention through a reversal of epigenetic aberrations.
Influence of Epigenetic Modifications on Cancers
Aging is associated with the increased development of most cancers. 54 Aberrant DNA modifications, including DNA hypomethylation of open sea regions and DNA hypermethylation of promoter CpG islands, have been observed in most of the cancers. 54 DNA hypermethylation on the promoter region of tumor suppressor genes, including Rb, p14, p15, p16, p21, TIGI, RUNX3, and regulatory genes such as RAS association domain family 1A (RASSF1A) and retinoic acid receptor β (RARB), DNA repair mechanism genes BRCA1 and MGMT can lead to their inactivation, genetic instability, and subsequent cancer development. 55 On the contrary DNA hypomethylation is associated with the activation of oncogenes and tumor formation. 55 While DNA hypermethylation is more common in cancers, DNA hypomethylation is also seen in various types of tumor, including cervical, ovarian, prostate, liver, and B cell chronic lymphocytic leukemia. 55
Aberrant histone modifications are also observed in many cancer types. It was found that cancer cells exhibit downregulation of histone acetylation at H4K16 and loss of histone methylation at H4K20me3. 56 HDAC1 and HDAC2 expression was found to be increased in prostate and gastric cancers, respectively. 57,58 A deletion of EZH2 (H3K27 methyltransferase) results in the increased development of spontaneous T cell leukemia. 59 Chen et al. found that low levels of H3K4ac and elevated expression of H3K27me3 were associated with the progression of oral squamous cell carcinoma. 60
EZH2, SUZ12, and EED subunits are the components of methyltransferase Polycomb Repressive Complex 2 (PRC2) and are predicted to the valid targets for tumorigenesis through histone modification at H3 on lysine 27 residue (H3K27me3). 61 PRC2 inhibitors (MAK683, EED226, and FDA-approved EPZ6438) reduced the proliferation of lymphoma cell WSU-DLCL2 (WSU), rhabdoid tumor (MRT) cells, pancreatic cancer cell Hs700T with SMARCB1 deficiency, and ovarian cancer cell A2780 with ARID1A deficiency through altering the methylation and acetylation of H3K27 of ECM, SASP, proteoglycan, and cell cycle genes, including GATA4, MMP2/10, ITGA2, and GBP1, as well as cell cycle regulatory gene CDKN2A/p1661 suggesting chromatin modification as a key upstream pathway in balancing the cell cycle for controlling proliferation under stress conditions.
However, these PRC2 inhibitors failed to reduce the proliferation of refractory RD, HeLa, and mast cell activation syndrome (MCAS cells) although affecting the methylation process 61 hinting at the differential mechanism of chromatin remodeling in cervical cancer and other immune-related disorders. Another study reported that dual inhibition of H3K9 and H3K27 methylation led to reduced metastasis and proliferation of tumor cells such as breast cancer (MCF7, BT549, and MDA-MB-231), colon cancer (SW480 and HCT116), and prostate cancer (PC3 and DU145) using G9a inhibitor (UNC0642) and the EZH2 inhibitor (UNC1999, or UNC0642 + UNC1999). 62 Chromobox family proteins (CBX2/3/8) were found upregulated in glioblastoma suggesting the importance of epigenetic reprogramming in controlling the disease and disorders. 63 These findings indicate that aberrant DNA methylation of tumor suppressors and oncogenes and H3K27 methylation are associated with the development of most of the cancers.
Influence of Epigenetic Modifications on Senescence and Telomere During Aging
Senescence and aging cannot be used interchangeably but the number of senescent cells increases during aging and contribute to age-related dysfunction by reducing the regenerative capacity of tissues. 64 Decreased expression of histone methyl transferase nuclear receptor binding SET domain protein 2 (NSD2) was observed in the senescent bone marrow MSCs (BMMSCs), which suggests that epigenetic alteration in tissue-resident stem cells can contribute to aging phenotype by driving them toward senescence and reducing their regenerative property. 65 Giuliani et al., observed genome-wide DNA hypomethylation in replicative senescent BMSCs and human umbilical vein endothelial cells, which is associated with different pathways, including Wnt/β-catenin signaling, molecular adhesion, and insulin resistance. 66 Zhang et al. reported an increase in genome-wide H3K9 acetylation in flag leaf senescence suggesting its role in the sexual reproduction of cereal crops, crop growth, and reducing photosynthesis and grain production. 67
It was shown that KDM4A-mediated H3K9me3 and DNMT3B promote senescence in NPCs leading to intervertebral disc degeneration. 68 Histone demethylase KDM4B ablation is also known to cause a defect in the self-renewal of MSCs by promoting senescence-associated heterochromatin foci leading to skeletal aging. 69 Moreover, replicative senescence is demonstrated as the causative agent of aging in several cell types. Li et al. observed decreased expression of Ezh2 (Enhancer of zeste homolog 2) in the fibrotic left atrium tissue of aged mice and senescent atrial fibroblast due to replicative senescence through modulating the H3K27me3 level in the promoter region of CDKN2a (p16, p19) and Timp4 gene suggesting the role of histone marks in aging by regulating replicating senescence. 70 A study reported maturation phase transient reprogramming where dermal fibroblast from middle-aged donors acquired their fibroblast identity possibly through the rejuvenation of the DNA methylation aging clock suggesting full reprogramming is not required to reverse the aging of somatic cells. 71
Ezh2 expression was also found to be associated with increased cellular senescence in Ang II-induced vascular smooth muscle cell aging model through regulating H3K27me3 levels at ANXA6 promoter. 72 Histone H3 lysine 4 (H3K4) methyltransferase Smyd3 and elevated H3K4me3 modification were reported to promote senescence in rat endothelial cells. 73 These research reports advocate that H3K4me3, H3K9me9, and H3K27me3 histone marks are associated with senescence and can be used as possible therapeutic targets to rejuvenate aging cells and tissue.
Senescence and aging are also induced by telomere shortening. Telomerase is a ribonucleoprotein enzyme important for telomere elongation. 74 Telomerase is expressed at low levels in somatic cells and is constitutively expressed in stem cells and germ cells. However, telomerase activity was shown to be insufficient in these cells to maintain normal telomere length and thus gradual shortening of telomeres occurs over time and leads to replicative senescence, which may contribute to aging and age-related disorders. 74 Takasawa et al. observed increased methylation in telomerase reverse transcriptase (TERT) promoter differential methylated region of iPSCs during reprogramming, which is associated with its increased promoter activity. Furthermore, they noted decreased methylation in the TERT promoter region of parental somatic cells, which also corresponds to generally low expression of telomerase in them. 75 This study indicates that the methylation status of TERT is important for its expression, thus rendering telomerase susceptible to epigenetic changes during aging and its expression.
It was found that overexpression of telomerase can extend the lifespan of mice indicating its antiaging potential. 76 Another study demonstrated decreased expression of telomerase and relative telomere length in Nipponia nippon, Colombia livia, Pelodiscus sinensis, and Xenopus laevis with age while no correlation was found in Alligator sinensis, which indicates that telomerase and telomere shortening is not a single parameter to assess age. 77 It was found that DNAmGrimAge and DNAmPhenoAge acceleration was inversely related to the length of the telomere suggesting the degree of methylation at the telomere drive telomere shortening during aging. 78 A study observed increased H3K4 and H3K79 methylation at telomere-proximal regions of replicative aged yeast cells suggesting that silencing of subtelomeric regions may have an impact on telomere length. 79 However, it is unknown whether epigenetic modification at the telomeric or subtelomeric end has any effect on the regulation of telomerase expression.
Telomere length is also known to regulate the activity of human telomerase catalytic subunit hTERT by modulating the DNA methylation and histone marks on its promoter region during aging. 80 Kim et al. observed that aged BJ human fibroblast cells with short telomeres exhibit increased expression of two active marks, H3K4me3 and H3K9ac, and one repressive mark, H3K27me3, in TERT promoter as compared with young cells with long telomeres. Collectively, these results indicate that the hTERT promoter is more permissive in aged cells suggesting hTERT expression is influenced by the length of telomere and histone modifications. 80 Taken together, these data propound that telomerase can be used as an antiaging marker by modulating its TERT promoter methylation status using epigenetic modifiers.
Methods for the Detection of DNA Methylation
DNA methylation commonly occurs at 5 methylcytosine within CpG residues, which plays an important role in the regulation of gene expression, cell proliferation, differentiation, and genomic stability. 81 The reversible nature of DNA methylation demands an effective method for its diagnosis and treatment. 81 The various methods to detect DNA methylation changes are described below. Figure 1 depicts the comparison between these methods.
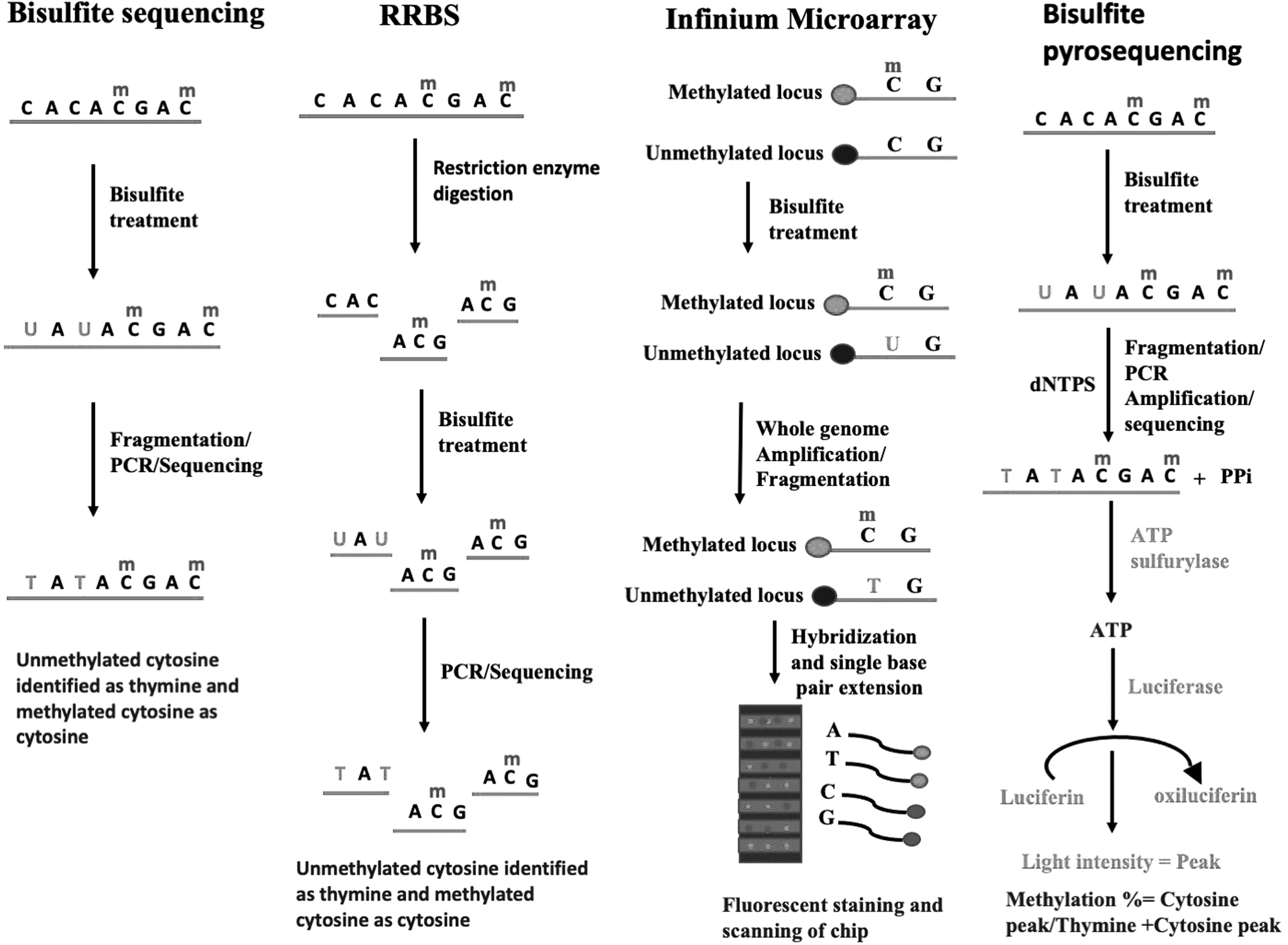
Methods for the detection of DNA methylation.
Bisulfite Genomic Sequencing
Bisulfite genomic sequencing is considered a gold standard method for the detection of DNA methylation at CpG sites at single base-pair resolution. 81 In this method, sodium bisulfite treatment of DNA results in the deamination of cytosine to uracil in unmethylated DNA, which gets converted to thymine in subsequent PCR amplification and detected by sequencing. 81 However, DNA methylation at 5 methyl cytosine remains unaffected by this conversion and is recognized as cytosine in subsequent PCR amplification and detection thus allowing the distinction between methylated and nonmethylated DNA. 81
Reduced Representation Bisulfite Sequencing
In this method, one or more restriction enzyme such as MsPI is used to cut the DNA to generate sequence-specific fragments and analyze DNA methylation at single nucleotide resolution. 82,83 MsPI digestion cuts at CCGG sites and recognizes both methylated and nonmethylated sites that result in the isolation of 85% of CpG islands. 82 The fragmented DNA is then treated with sodium bisulfite and sequenced. 82,83 This method is effective where methylation level is high such as repeat regions and promoters. 83
Infinium Human Methylation BeadChip Design
Illumina has developed cost-effective and high-throughput Infinium humanmethylation microarray assay for the detection of methylation across the genome. 84 These human methylation-based array platforms include HumanMethylation27 (27 K) BeadChip, Illumina Human Methylation 450 K (450 K) array, and Infinium MethylationEPIC (EPIC) BeadChips which detects DNA methylation at single base resolution through measuring sodium bisulfite-treated DNA. 84,85 These arrays are based on Illumina's BeadChip technology and do not depend on polymerase chain reaction (PCR) and detect signals based on dye intensity. 86 In these arrays, DNA is first treated with sodium bisulfite and then subjected to whole genome amplification, fragmentation, hybridization, and single base-pair extension on the microarray chip. 84
In the 27 K BeadChip, CpG site detection is based on two 50 bp probes produced by two different bead types to hybridize methylated CpG site (M) and unmethylated CpG site (U) and generate the same color signal for both M and U while 450 K BeadChip produces signal using two different probes (Illumina Infinium I and II) and employs two different color green and red to differentiate between M and U signals. 84 HumanMethylation27 (27 K) BeadChip can assess 27,578 CpG sites, including 14,495 protein-coding gene promoters while HumanMethylation450 (450 K) BeadChip assays can interrogate 482,421 CpG sites and Infinium MethylationEPIC (EPIC) BeadChips can detect 935,000 CpG sites across epigenome. 84,87 For Illumina BeadChip arrays, methylation levels are calculated as beta value β = M/(M + U + α), where α arbitrary value is usually 100. 84 Moreover, the Epic array detects only 30% of epigenome rendering a large number of CpG sites unmeasured. 86
Another method known as methylation Capture bisulfite sequencing (MC-seq) can detect 3,708,550 CpG sites at single nucleotide resolution with more CpGs in the coding region and CpG islands at an affordable price utilizing a targeted next-generation sequencing approach. 86 Whole-genome bisulfite sequencing is a next-generation sequencing technique and can annotate more than 28 million CpG sites, but is associated with the high cost and large input genomic DNA requirement. 86
Bisulfite Pyrosequencing
Bisulfite pyrosequencing is a quantitative method to determine the methylation status of bisulfite-treated CpG sites at a single nucleotide level. 88 This method is regarded as the gold standard for the measurement of allele-specific methylation patterns. 89 In this method, DNA is first treated with bisulfite to generate either cytosine or thymine for the identification of methylated and nonmethylated cytosines. 88 Furthermore, it involves the addition of deoxynucleotide triphosphate to the growing strand of DNA, which results in the release of pyrophosphate (PPi) that gets subsequently converted to ATP by an enzyme ATP sulfurylase. 88,89 The ATP is utilized by the luciferase to convert the luciferin to oxyluciferin. 88,89 The amount of light produced is detected and recorded as a peak, which is proportional to the amount of nucleotide incorporated. 88,89 The methylation percentage is calculated as the ratio of the cytosine peak to the sum of the thymine and cytosine peaks. 89 The main disadvantage of this method is that it analyses only smaller regions (350 bp). 89
Epigenetic Aging Clock
The epigenetic clock can be used as a diagnostic marker for the prediction of biological age. These biomarkers can be measured within tissues or cells or body fluid and thus can be used for disease detection or prognosis or therapy monitoring. 90 In 2013, Horvath analyzed ∼8000 samples from 51 healthy human tissues and cell types as well as ∼6000 cancer samples and revealed exclusive 353 CpGs methylation sites called clock CpGs (also known as pan-tissue Horvath DNAm clock) accurately predicting age. 91,92 Parallelly, Hannum et al. identified 71 CpGs sites (also known as Hannum DNAm clock) to predict aging with high fidelity from the blood samples collected from individuals 19–101 years of age. This clock can be utilized to measure the aging rate in age-related diseases such as tumors. 93 Furthermore, Horvath laboratory developed the DNA methylation PhenoAge clock (also known as Levine clock) based on 513 CpG sites for the measurement of aging and diseases such as cardiovascular disorder using DNA methylation values from whole blood. 94
Later, the same group released the DNA methylation GrimAge clock, which is reported to predict mortality, coronary heart disease, and cancer. 95 Recently, Milicic et al., reported an association between age acceleration and hippocampal volume within Amyloid-β-positive (Aβ) individuals using Hannum, Zhang, and Phenoage epigenetic clocks. It is well known that Aβ plaques is the main cause of AD and accumulates with aging. 96 In addition, several groups generated epigenetic aging clocks in mice. 97 –101 Furthermore, Knight et al. generated an epigenetic clock based on 148 CpG sites from cord blood to estimate the gestational age at birth. 102 Recently, McEwen et al. has identified Pediatric-Buccal-Epigenetic clock based on 94 CpG sites from buccal epithelial cells for measuring age in the pediatric population. 103 More recently, an epigenetic clock based on ribosomal DNA is discovered and it is found to be evolutionarily conserved in different species. 104
Few reports are suggesting the cost-effective generation of an epigenetic age clock based on a few CpG sites by bisulfite pyrosequencing method suggesting the feasibility of age estimation by a few biomarkers. 105,106 Earlier, Weider et al. developed a cost-effective epigenetic clock based on three CpG sites related to aging genes ASPA, ITGA2B, and PDE4C from human blood. 105 Later, Han et al. also generated an epigenetic clock by bisulfite pyrosequencing method in mice blood-based on three CpG sites in genes Hsf4, Prima1, and Kcns1. 106
Recently, two dual-species human–opossum pan-tissue clock was developed to accurately measure chronological and relative age, respectively, in human and opossum. 107 It was reported that higher GrimAge is a predictor of old-age cognitive function and was shown to be associated with reduced cognitive and brain vascular age during later stages of life. 108 Sugden et al. observed that first-generation clocks, including Horvath and Hannum clocks, and second-generation clocks, including GrimAge and PhenoAge, were not effective in measuring the cognitive impairment in older adults while third-generation measurement of biological aging using DunedinPACE was able to diagnose AD and risk of development of dementia. 109 A recent study using Horvath's clock demonstrates that higher body mass index (BMI) is associated with age acceleration in visceral adipose tissue but not in blood, further suggesting that obesity leads to age acceleration in metabolic active tissues. 110
Caulton et al. developed a farm animal epigenetic clock using the “HorvathMammalMethyl40” methylation array for predicting the age of livestock animals, including goats, cattle, Red and Wapiti deer, and composite-breed sheep. 111 Using Illumina HumanMethylationEPIC BeadChip, it was found that an increase in systolic blood pressure and pulse pressure accelerated epigenetic age. 112 Recently, Lam et al. developed epigenetic MRI (eMRI) that can measure DNA methylation changes in the living human brain paving the way to identify the onset of aberrant methylation and remedy for same. 113 Manco et al. developed duplex droplet digital PCR (ddPCR) assay to measure DNA methylation changes in the ELOVL2 gene as a biomarker for age prediction in forensic science. 114 These epigenetic biomarker clocks provide a platform to understand the epigenetic alteration during development, aging, and age-related disorders and their comparison among species.
These studies showed that most of the epigenetic clocks are generated by utilizing reduced representation bisulfite sequencing (RRBS) or Illumina Infinium HumanMethylation450 BeadChip assay methods. HumanMethylation450 (450 K) BeadChip assays is recently been replaced by Infinium MethylationEPIC (EPIC) BeadChips array due to the doubling of the number of CpG sites. 115 However, various studies have reported that a subset of 450 K CpG sites is absent in Epic arrays thus raising the utility of EPIC arrays to accurately estimate the biological age. 115,116 Hence, further studies are needed to test the efficacy of EPIC array to measure the biological age. Table 1 represents the method used by different studies for biological age estimation. Recently, it was shown that the DNA hypermethylation profile of blood (108 markers) and frontal cortex (514 markers) depicted linear relation with aging (8–96), whereas cerebellum (137 markers) showed nonlinear relation with aging due to the saturation of DNA methylation as age increased suggesting underestimation of age using cerebellum sample. 117
Methods for Development of Epigenetic Clock and Measurement of Biological Aging
Pavanello et al. observed that the DNAmAge of the right and left kidney is older than blood DNAmAge and age acceleration is faster in the right and left kidney than in blood from the same donor. Furthermore, they noted that the telomere length of the right and left kidneys is longer than blood. 118 Therefore, further research is required for dissecting the correct tissues for age estimation under normal and disease conditions.
Epigenetic Aging Clock for Cancer
Aging and cancer hold a complex relationship. Recent studies have been developed to measure biological aging and predict cancer risk based on DNA methylation sites known as epigenetic clock and epigenetic age acceleration. 119 Dugue et al. assessed the association between age acceleration and various cancers using Human Methylation 450 K BeadChip assay. They observed that epigenetic aging was associated with an increased risk of cancer with a 5-year age acceleration leading to 4%–9% cancer risk. 119 A study measured baseline blood DNA methylation of 2764 women and predicted age acceleration using three epigenetic clocks, including Horvath, Hannum, and Levine. They found that 5-year age acceleration was significantly associated with the development of increased breast cancer risk. Furthermore, they noted using Levine's clock that 5 years of age acceleration corresponded to a 15% increase in the development of breast cancer. 120 Using HumanMethylation450 BeadChips it was found that GrimAgeAccel was weakly associated with invasive breast cancer development mostly in postmenopausal women. 121
Durso et al. analyzed a publicly available leukocyte methylation large dataset to demarcate the relationship between epigenetic age and cancer development. Using epigenetic clocks, including Horvath, Hannum, and Weidner, and two CpG-specific ELOV2 and FHL2, they observed a positive correlation between DNA methylation and breast and colorectal cancer, respectively. 122 Zheng et al. found using the Horvath clock that epigenetic age acceleration is a predictor of colorectal cancer development, mortality, age, and tumor stage. 123 Chen et al., observed using Horvath, Hannum, and Levine clocks that DNA methylation-based biological age is positively associated with a risk factor for breast cancer, including BMI and alcohol. 124
A study analyzed 6000 cancer samples from 32 datasets and observed that some cancers showed a positive correlation with DNA methylation age, whereas others showed a negative correlation. 93,94 Using EAA Horvath and Hannum epigenetic clocks, novel genetic variants in the SELP gene and HLA region were found to associate with age acceleration in childhood cancer indicating the potential drug target for preventing cancer development. 125 These investigations demonstrate that DNA methylation age acceleration is associated with the development and progression of various tumors and can be used as predictive markers for various cancers.
Epigenome-Targeted Therapies
The emerging area of therapeutic intervention for the treatment of various diseases and disorders includes epigenetic drugs also known as “epidrugs”. 126 These epigenetic drugs are in different phases of clinical trials for the treatment of various disease conditions. They mostly act as inhibitors for DNA methyltransferases, histone-modifying enzymes, such as HMT, histone demethylases, HATs, HDACs, and protein arginine methyltransferases, while in some contexts they also act as activators. They act on the 3D architecture of chromatin encompassing a large network of signaling and metabolic pathways. 126 Recent research on epigenetic modifications has led to the development of new inhibitors, such as HMT, bromodomain and extra-terminal protein, lysine-specific demethylase 1A (LSD1/KDM1A), and protein arginine methyltransferase in addition to DNMT inhibitors and HDACi. 127 These epigenetic drugs are reported as potential therapeutics for the treatment of various types of cancers such as skin, blood, colon, breast, and prostate. 128
Several epigenetic drugs have been approved by FDA, while many of them are in preclinical and different clinical stages.
The DNA epigenetic modifiers include azacytidine, decitabine, EPG, SGI-110, DZNep, JQ1, EPG, and curcumin while azacytidine and decitabine are FDA approved. The five currently FDA-approved histone epigenetic modifiers are vorinostat (SAHA), VPA, romidepsin, belinostat, and panobinostat. Most of these drugs are either DNMT inhibitors or HDACi. 128 These epigenetic compounds are known to exhibit immunomodulatory, cytotoxicity, antigrowth, and apoptotic effects either alone or in combination on different cancer cells. 128 In particular, these drugs inhibit the vital signaling molecules, which are important for the reactivation of tumor suppressor genes and inhibition of the oncogenes; significant for tumor development and progression.
Many epidrugs have been identified for the treatment of cardiac hypertrophy and heart failure and one promising candidate is HDAC. Various HDACi are available for preventing and improving cardiac functions such as trichostatin A, scriptaid, suberoylanilide hydroxamic acid (SAHA, also known as Vorinostat), and SK-7041. These epigenetic drugs are involved in the suppression of cardiac hypertrophy, and fibrosis, reducing myocardial infarct size and preserving systolic function. 129 –131 Givinostat, another pan-HDACi is associated with reduced inflammatory response, angiogenic effects, and cardiac fibrosis. 132 DNA methylation inhibitor, 5-azacytidine also implicated in T2D and cancer, plays an important role in the improvement of cardiac function and cardiac fibrosis. 133 It can be inferred from these studies that these epigenetic modifiers can be used for reversing the epigenetic aberrations of age and age-related disorders. Table 2 represents the epidrugs for different age-related pathologies.
Epidrugs for Age Related Pathologies
Epigenetic Effect of Nutrition on Aging Reversal
Nutrition is also linked to epigenetic modification and is known to impact the aging rate. It is essential to understand the interaction between epigenetics, nutrition, and aging to unravel the factors, including diet, caloric restriction, methionine, and vitamins essential for promoting quality of life during aging. A study has shown that mice and monkeys who are exposed to caloric restriction from the early phase of their life result in a reduction of the age-related methylation drift, which ensues at a younger biological age than their actual chronological age. It was found that age-related hypermethylation is more prominent in CpG islands, whereas age-associated hypomethylation occurs at non-CpG sites. 134 In addition to this, other studies have also shown the association between caloric restriction and longevity through epigenetic modifications. 98,99,101 Minteer et al. observed that caloric restriction can slow down the epigenetic aging process and identified EED and Polycomb group as important chromatin regulators for cultural aging through reprogramming of lung and kidney fibroblast to induced pluripotent stem cells (iPSCs). 135
A recent report stated that both dietary restriction and rapamycin treatment can modulate DNA methylation at an early age than normal-aged mice indicating that preventing the DNA methylation changes associated with aging at a younger age can increase longevity. 136 Gong et al., observed a decrease in the level of H3R2me2, H3K27me3, H3K79me3, and H4K20me2 in aged brain mice and their reappearance with dietary restriction and rapamycin. 137 These reports indicate a positive correlation between caloric restriction and longevity.
Methionine is the important precursor for S-adenosylmethionine (SAM), which in turn gets converted to S-adenosylhomocysteine (SAH) by Dnmt. 138 The ratio of SAM/SAH is known as the methylation index whose level regulates DNA methylation reaction by donating methyl group to CpG island. 138 Various reports suggested that supplementation of methionine resulted in DNA hypermethylation and decreased gene expression. 138 Several reports state that SAM/SAH ratio alters histone methylation, including H3K4me3, H3K36me3, and H3K79me3 marks in yeast, C-elegans, and drosophila model systems. 139 Various studies have shown that methionine restriction or dietary restriction is associated with increased lifespan in various animal models, including drosophila, c-elegans, mice, and rat through modulating DNA and histone modifications. 139 Methionine can be obtained from various food sources, including lamb, fish, beef, pork, egg, seeds, legumes, cereals, vegetables, and fruits 140 indicating that a methionine-rich diet has the potential to regulate DNA methylation reaction and thus can play a role in delaying or reversing aging or age-related disease conditions.
However, it is unclear how aging influences methionine metabolism in different tissues and organs. It is important to understand how methionine affects the metabolic flux with age and influences aging and age-related disorders. Moreover, further work is required to understand the influence of methionine on various methyltransferases and identify specific methyltransferases affected by the same. Thus, additional studies are needed to study the role of methionine as an epigenetic modifier in reversing/delaying aging or age-related disorders.
Folate is another metabolite whose supplementation positively correlates with SAM concentration and has a well-documented role in DNA methylation and less for histone methylation. 141 It is known to bind LSD1 demethylase of H3K4me1/2 preventing its inhibition by formaldehyde thus suggesting its role as an epigenetic modifier. 142 A study has reported that folic acid and vitamin B12 supplementation results in global hypermethylation in the unmethylated regions of CpG islands in 44 participants, whereas monomeric and oligomeric flavanols result in hypomethylation in 13 participants. They further found a reduction in the Horvath “epigenetic clock” with the MTHFR 677CC genotype in women following folic acid and vitamin-12 supplements. 143 It would be interesting to study the influence of the combinational effect of various nutritional epigenetic modifiers in increasing the age of various organisms.
Recently, Gensous et al. has demonstrated that a 1-year intake of a Mediterranean-like diet leads to epigenetic rejuvenation in elderly individuals with country, sex, and individual-specific effects. 144 Cavallucci et al. observed that β-Hydroxy-β-Methyl Butyrate increases global histone acetylation, whereas β-Hydroxybutyrate increased lysine β-hydroxybutyrylation (Kbhb) of histone tails in muscle cells highlighting that these compounds can be tested for aging reversal. 145 A study revealed that soy-based food component genistein affected the binding properties of various epigenetic regulators, including ATRX, SUV39H1/H2, and HP1BP3 resulting in reduced growth of cancer cells through the downregulation of proliferating genes. 146 It was found that the combined effect of dietary phytochemicals, including sulforaphane (SFN), sodium butyrate (NaB), and Genistein has a more profound effect in downregulating HDACs (HDAC1, HDAC6, and HDAC11), DNMTs (DNMT3A and DNMT3B), HMT (EZH2 and SUV39H1), and HATs (GCN5, PCAF, P300, and CBP) and arresting the growth of breast cancer lines MDA-MB-231 and MCF-7. 147
A recent study showed that polyamine metabolism influences DNA methylation activity and a longer intake of spermidine increases the longevity of mice. 148 These investigations indicate that diet management could be an alternative strategy to prevent/reverse epigenetic modification during age-related disease conditions. However, it is yet to determine whether all epigenetic biomarkers reverse upon diet intake and slow down the progression of aging and age-associated disease conditions such as cancer, cardiovascular, diabetes, and neuronal disorders.
MSC Exosomes for the Treatment of Aging and Age-Related Disorders
MSCs and their paracrine secretion hold great value for the treatment of aging and age-related disorders. Sanz-Ros et al. observed that adipose MSC-derived extracellular vesicles were able to ameliorate various parameters associated with aging, including renal function, fatigue resistance, motor coordination, grip strength, and fur regeneration, as well as able to decrease inflammation, oxidative stress, and senescent markers in muscle and kidney. 149 Various studies have shown that MSCs and their exosomes were able to slow brain aging by reducing/suppressing the secretion of inflammatory regulators, promoting angiogenesis, and neuronal cell repair. 150 A study found that coculturing IMR-90 senescent cells with MSCs was able to reduce the expression of interleukin 6 (IL-6) and reverse the expression of various growth factors, including growth differentiation factor (GDF11) and TGFβ1 as well as increased the number of mitochondria and telomere length. Moreover, they also observed that MSCs were able to reduce the symptoms related to aging in aged mice. 151
Skin aging is characterized by a decrease in the expression of extracellular matrix components, including elastin and hyaluronic acid, which leads to poor skin turgor and elasticity. Ong et al. found that condition media (CM) derived from red deer umbilical cord MSCs was able to upregulate the expression of hyaluronic acid by 83% and elastin by 56% in human dermal fibroblast suggesting the beneficial effect of CM on skin aging. 152 MSCs and their secretome are also known to alleviate age-related disease conditions, including diabetic wound healing, immune dysfunction, bone repair and knee osteoarthritis through secreting various growth factors, and extracellular matrix components and inhibiting oxidative stress and inflammatory molecules. 153 –156 Various studies have shown that the rejuvenation of aged stem cells can be mediated by the secretome from young MSCs. 157 However, the mechanism underlying the same is unclear.
Epigenetic modifications provide novel therapeutic targets for the treatment of aging and age-related disorders. It is yet to find out whether MSCs mediate their action in ameliorating aging and age-related diseases and disorders through modulating epigenetic mechanism. Recently, we hypothesized whether MSCs can act as epigenetic modifiers upon systemic or local transplantation. 158 MSC exosomes constitute various growth factors and miRNAs suggesting their suitability as epigenetic modifiers. 159 It would be worthwhile to check if it is possible to manipulate MSC secretome for epigenetic modification through genomic modification and use as cell therapy to rejuvenate cells, tissues, and organs. Moreover, MSC exosomes can be used to replenish the lost miRNAs or epigenetic modifying enzymes or their activators/factors during aging through genomic modifications. Since MSCs are also prone to undergo aging under adverse conditions, it would be essential to use young MSCs CM/exosomes as epigenetic modifiers rather than MSC transplantation.
Conclusion and Future Direction
Research over the last decade has greatly increased our knowledge about the epigenetic mechanism underlying aging using various model systems, including drosophila, c-elegans, yeast, mouse, and humans (Table 3). These studies indicate epigenetic variance among species during aging suggesting that epigenetic alterations need to be closely monitored in humans for the development of therapeutic targets for slowing or reversing aging. iPSCs derived from the aged individual can be used as a human model system to identify epigenetic alteration and for the screening of epidrugs to reverse the epigenome aberrations.
Epigenetic Modifications Among Different Species During Aging
It is yet to prove if it is possible to demarcate aging and different aging-related abnormalities based on epigenetic marks. So far, these studies show different age-related abnormalities harbor different epigenetic marks and can be distinguished based on different histone marks and epigenetic clocks (Table 4). These studies have shown that H3K9me3 is the most altered histone mark in various age-related pathologies. However, these studies also raise the possibility that epigenetic alteration may play a different role in different tissues and organs during aging. Hence, it is essential to study the epigenetic modifications in different tissues and organs during aging and the mechanism underlying the same for the development of therapeutic targets.
Epigenetic Modifications During Age-Related Pathologies
It is also vital to understand that normal aging and pathological aging also share common epigenetic alterations. For example, normal aging is associated with DNA hypomethylation whereas cancer is also associated with DNA hypomethylation, which suggests that there may be other factors, including lifestyle or environmental, which predispose the organism toward pathological aging. There could also be a possibility that extent of epigenetic alteration in whole tissue or organism may also be responsible for predisposing the organism toward pathological aging. Hence there is a vital need to understand all the molecular pathways influenced by an epigenetic mechanism under normal and diseased conditions during aging in particular tissue or organisms. From the foregoing account, it is clear that epigenetic changes initiate the aging process. Hence, the reversal of such changes is likely to halt the progression of aging and age-related disorders. However, it is not yet clear what causes the loss of epigenetic information during aging.
It is yet to prove whether epigenetic alteration during aging is a cause or consequence of aging. Yang et al. showed that transient induction of genomic instability through nonmutagenic DNA breaks results in the alteration of chromatin changes, including loss of epigenetic information and cellular identity along with the acceleration of epigenetic clock and cellular senescence suggesting epigenetic aberration is a cause of aging in mammals. 160 High-throughput sequencing, single-cell genomics, and CRISPR technology will greatly enhance our understanding of identifying the key epigenetic manipulations and mutations in epigenetic enzymes/factors that are casuals of aging and age-related disorders. Identification of new epigenetic modifiers to reverse aging and age-related abnormalities and to what extent/fast these epigenetic modifications can be reversed would pave a way for a healthy and longer lifespan. Therefore, the future of epigenetic therapy lies in the exploration of compounds that can act as epigenetic modifiers. MSC exosomes exhibit the potential in reversing various related abnormalities, however, the mechanism is unclear.
We have proposed the use of MSC exosomes as epigenetic modifiers. MSC exosomes harbor various growth factors and miRNAs describing their potential as epigenetic modifiers. MSCs can be genetically modified to produce exosomes that can serve as cargo to replenish the lost miRNAs or factors or DNA and histone-modifying enzymes during aging. Other factors that can act as epigenetic modifiers include diet, methionine, folic acid, and vitamins and bioactive compounds. It would be interesting to explore if epigenetic clocks, which are set of CpG sites can estimate the influence of various epigenetic modifiers, including methionine, folic acid, caloric restriction, diet, and MSC exosomes on increasing the lifespan of various organisms. In addition, various studies suggest the efficiency of Yamanaka reprogramming factors for reversing the age-induced epigenetic changes in aged hematopoietic stem and progenitor cells, human senescent and centenarian fibroblasts, as well as the fibroblast isolated from HGPS patients. 161
Interestingly, Strollo et al. (2018) have revealed that astronauts coming back from long-term space missions showed health problems, which closely resemble the old age population indicating that aging might be faster in space than on earth. 162 However, it remains to be seen to what extent aging can be halted on earth as well as on spacecraft by reversing epigenetic signatures.
Footnotes
Acknowledgments
S.S. and R.B. contributed to the concept, data collection, and analysis of the article. S.S. wrote the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.