
Abstract
Smooth muscle cell (SMC) differentiation and dedifferentiation play a critical role in the pathogenesis of cardiovascular diseases. The lack of a good and simple in vitro SMC differentiation system has hampered the progress of SMC field for years. The generation of such an in vitro system would be invaluable for exploring molecular mechanisms of SMC differentiation and dedifferentiation. Recently, the establishment of induced pluripotent stem (iPS) cells has offered a novel therapeutic strategy to generate patient-specific stem cell lines. Here we have investigated whether iPS cells are able to differentiate into SMCs in vitro. Mouse iPS cell (O9 and TT025) monolayers were treated with 10−5 mol/L all-trans retinoid acid (RA). After 8 days of RA treatment, we found that >40% of the O9 iPS cells expressed the SMC-markers including SMα-actin and SM myosin heavy chain. Also, we documented that iPS-derived SMCs acquired SMC functional characteristics including contraction and calcium influx in response to stimuli. Moreover, our results indicated that there were differences in SMC-specific gene expression patterns between SMCs derived from O9 and TT025 iPS as well as normal embryonic stem cells. These differences might be due to disparity in the current iPS technology. Taken together, our data have established a simple iPS-SMC system to generate SMCs in vitro, which has tremendous potential to generate individualized SMCs for vascular tissue engineering and personalized drug screening.
Introduction
I
Materials and Methods
Cell culture
Mouse iPS cells (O9 and TT025, kindly provided by Dr. Jaenisch at the Massachusetts Institute of Technology) [3] and mouse ES cells (including B6-white, CMTI-1, and CMTI-2) were routinely cultured on tissue culture plates coated with 0.1% gelatin (Sigma-Aldrich, St. Louis, MO, USA) in Dulbecco's modified Eagle's medium (DMEM)/F12 (Invitrogen, Carlsbad, CA, USA) in the presence of 15% ES cell–qualified fetal bovine serum (Hyclone, Logan, UT, USA), 0.1 mmol/L 2-mercaptoethanol, 1 mmol/L
Immunocytochemistry
Derived cells were trypsinized with TrypLE™ Express for 5 min. Afterward, cells were resuspended in differentiation medium and plated on 0.1% gelatin-coated chamber polystyrene vessels (BD Bioscience, Bedford, MA, USA) and incubated in differentiation medium. Cells were then fixed and permeabilized using BD cytofix/cytoperm™ kit (BD Bioscience, Bedford, MA, USA), and incubated overnight with the primary antibodies, mouse anti-smooth muscle (SM)-α-actin 1:1,000 (Chemicon International, Temecula, CA) and rabbit anti-SM myosin heavy chain (SMMHC) 1:300 (Biomedical Technologies Inc., Stoughton, MA). After rinsing, secondary goat anti-mouse or rabbit immunoglobulin G (IgG) alexa fluor 594 (red) or 488 (green) 1:500 (Molecular Probes, Eugene, OR, USA) were added to the samples, which were then incubated for an additional hour. The cell nuclei were stained with 4′-6-Diamidino-2-phenylindole (Molecular Probes, Eugene, OR, USA). Finally, the cells were rinsed once more and mounted with mounting media. The slides were analyzed using fluorescence microscopy (Olympus, Japan). Normal mouse IgG1 and IgG2a (DAKO Corporation, Carpinteria, CA, USA) served as the negative control for SMMHC and SM-α-actin primary antibodies, respectively.
Fluorescence-activated cell-sorting analysis
After derived differentiated cells were resuspended in differentiation medium and centrifuged, the pellets were fixed and permeabilized using a BD cytofix/cytoperm™ kit and incubated overnight with the primary antibody. Mouse IgG1 or IgG2a served as isotypic controls, whereas 293 cells and rat vascular SMCs served as the negative and positive controls, respectively. Secondary goat anti-mouse IgG alexa fluor 488 (Molecular Probes, Eugene, OR, USA) was added to the samples, which were then incubated for an additional hour. Finally, the cells were rinsed once more and fluorescence was analyzed using the FACSCalibur™ system (BD Biosciences, San Jose, CA, USA) following the user's guide.
qRT-PCR
Total RNA from dimethyl sulfoxide (DMSO)- or RA-treated cells was extracted by using the RNeasy mini kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Complementary DNA (cDNA) was synthesized from 5 μg of total RNA with a superscript III first-strand synthesis system (Invitrogen, Carlsbad, CA, USA). cDNA samples were subjected to PCR amplification with primers specific for mouse SMC genes and other control genes. PCR primers and reaction conditions are described in Supplementary Table 1 (Supplementary Table is available online at
Western blot analysis
Protein samples were extracted using the M-PER mammalian protein extraction reagent (Pierce, Rockford, IL) supplemented with a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Antibodies against SM-α-actin (1:2,000 dilution, Chemicon International, Temecula, CA, USA), SMMHC (1:3,000 dilution, Biomedical Technologies Inc., Stoughton, MA, USA), and β-tubulin (1:10,000 dilution, Upstate, Charlottesville, VA, USA) were used for testing SMC-specific protein expression. Immunoactivity was visualized by the enhanced chemiluminescence's detection system (ECL, Amersham Biosciences, Pittsburg, PA, USA) according to the manufacturer's instructions.
Contractility assays
Agonist-induced contractile activity of the differentiated cells was assayed as previously described [13]. Derived differentiated cells with DMSO- or RA treatment were washed with phosphate-buffered saline (PBS), stimulated with 1 mmol/L carbachol (Sigma-Aldrich, St. Louis, MO, USA) in the differentiation medium and monitored under the microscope for 5 min. Images of the same field before and after carbachol treatment were snapped and compared.
Measurement of physiological responses
SMCs derived from mouse ES and iPS cells were cultured on a 35-mm circular coverslip and loaded with the calcium-sensitive fluorophore, Fluo-4 AM (2.5 μmmol/L; Molecular Probes, Eugene, OR, USA), and mounted into a constant-flow superfusion chamber as previously described [14]. Observation of intracellular Ca2+ changes was performed with confocal microscope (LSM 510 Meta, Carl Zeiss MicroImaging Inc, Thornwood, NY, USA). Fluo-4 was excited at 490 nm by a mercury arc lamp, and fluorescence emission was measured at 528 nm.
Statistical analysis
All data, calculated as a percentage from three independent experiments, were expressed as the mean ± SD and were evaluated with a χ2 test. A value of P < 0.05 was considered statistically significant.
Results
We have previously shown that human ES cells were able to differentiate into SMCs in response to the RA treatment using an EB-independent method [12]. In this study, we set out to determine whether iPS cells could also differentiate into SMCs in response to RA treatment in vitro. Mouse ES cells as well as iPS cells O9 and TT025 were induced to differentiate into SMCs following the protocol shown in Supplementary Fig. S1A (Supplementary Figure is available online at
O9 iPS cells treated with 10−5 mol/L RA for 8 days displayed SMMHC and SM-α-actin expression as determined by immunofluorescence (Fig. 1A), similar to the expression observed in mouse ES cells. Fluorescence-activated cell-sorting (FACS) analysis was performed with anti-SMMHC antibody to further assess the degree of homogeneity of SMC differentiation. As shown in Fig. 1B, SMMHC-positive cells significantly increased in both O9 iPS and ES cells after treatment with RA for 8 days, when compared to vehicle-treated spontaneous differentiation (O9 iPS cells: 41.26 ± 2.15% vs. 5.92 ± 0.94%, n = 3, P < 0.05, mouse ES cells: 50.33 ± 3.07% vs. 9.32 ± 1.02%). In parallel, western blot analyses demonstrated that expression levels of SM-α-actin and SMMHC proteins were upregulated in a time dependent-manner in RA-treated O9 iPS cells, although the final SMMHC levels was different between mouse ES and O9 iPS cells (Fig. 1C). To confirm the western blot data, the expression of SMC-specific genes was determined by qRT-PCR to demonstrate the effect of culture conditions on SMC differentiation (Fig. 2). A SMC-specific transcriptional coactivator (Myocardin), intermediate stage markers (SM22α and SM-α-actin), and a late stage marker (SMMHC) were significantly upregulated in RA-treated mouse ES and O9 iPS cells. Simultaneously, there was an obvious downregulation of markers of undifferentiated ES cells (Oct4 and Nanog) (data not shown). In addition, genes specifically expressed in cardiomyocytes (cTnI), endothelial cells (PECAM-1), and skeletal myocytes (MCKM) were maintained at low levels during this RA-induced SMC differentiation process (Supplementary Fig. S2). Moreover, endogenous or total expression level of Oct4, Klf4, Sox2, and c-Myc, which had been used for generation of iPS cell lines, were tested by using specific primers [2] during O9 iPS cell differentiation and showed that most transcripts of these four transcription factors originated from the endogenous locus rather than the viral vector, except c-Myc (Supplementary Fig. S3).
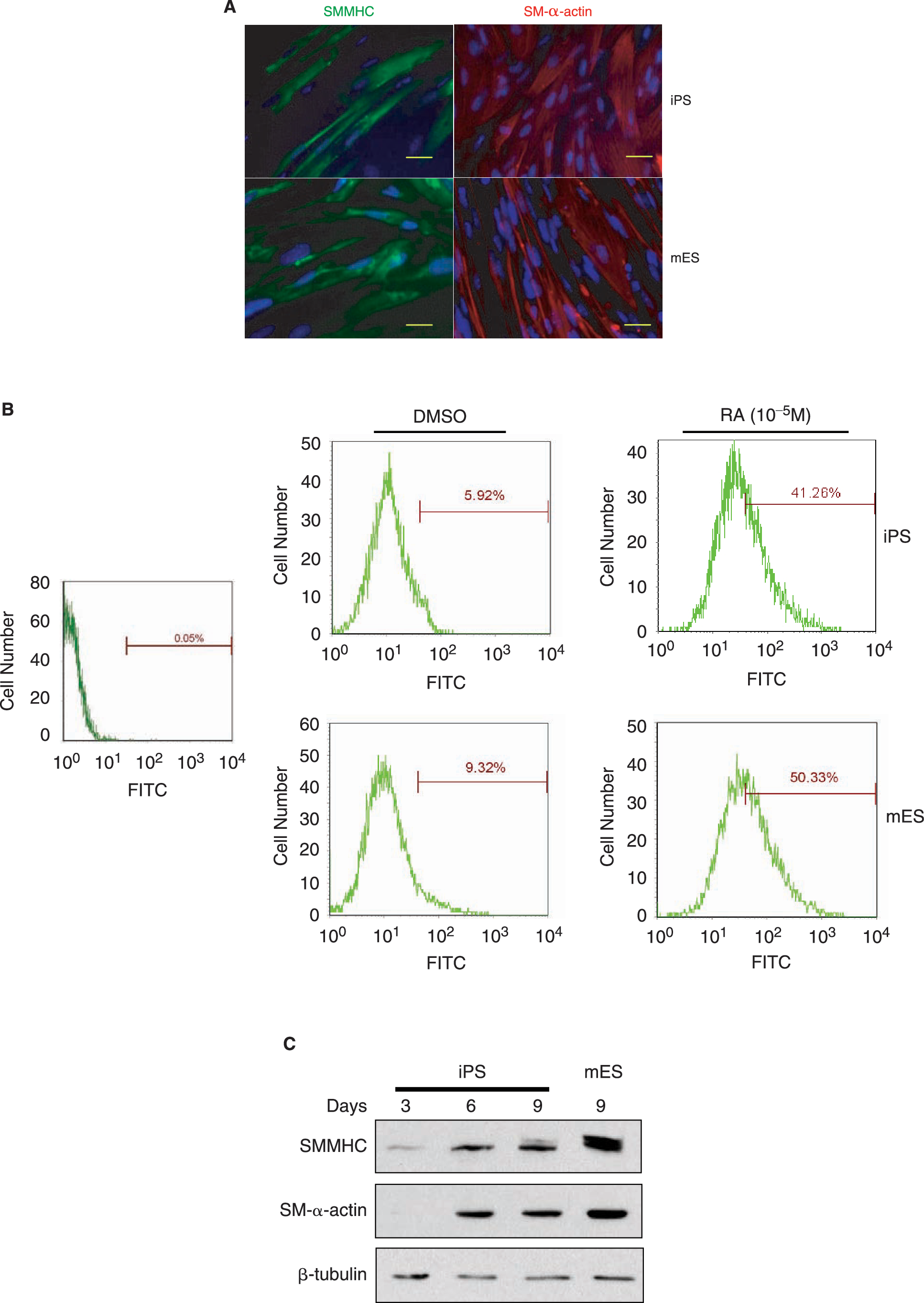
Mouse iPS cells can be induced to SMC differentiation by RA treatment. (
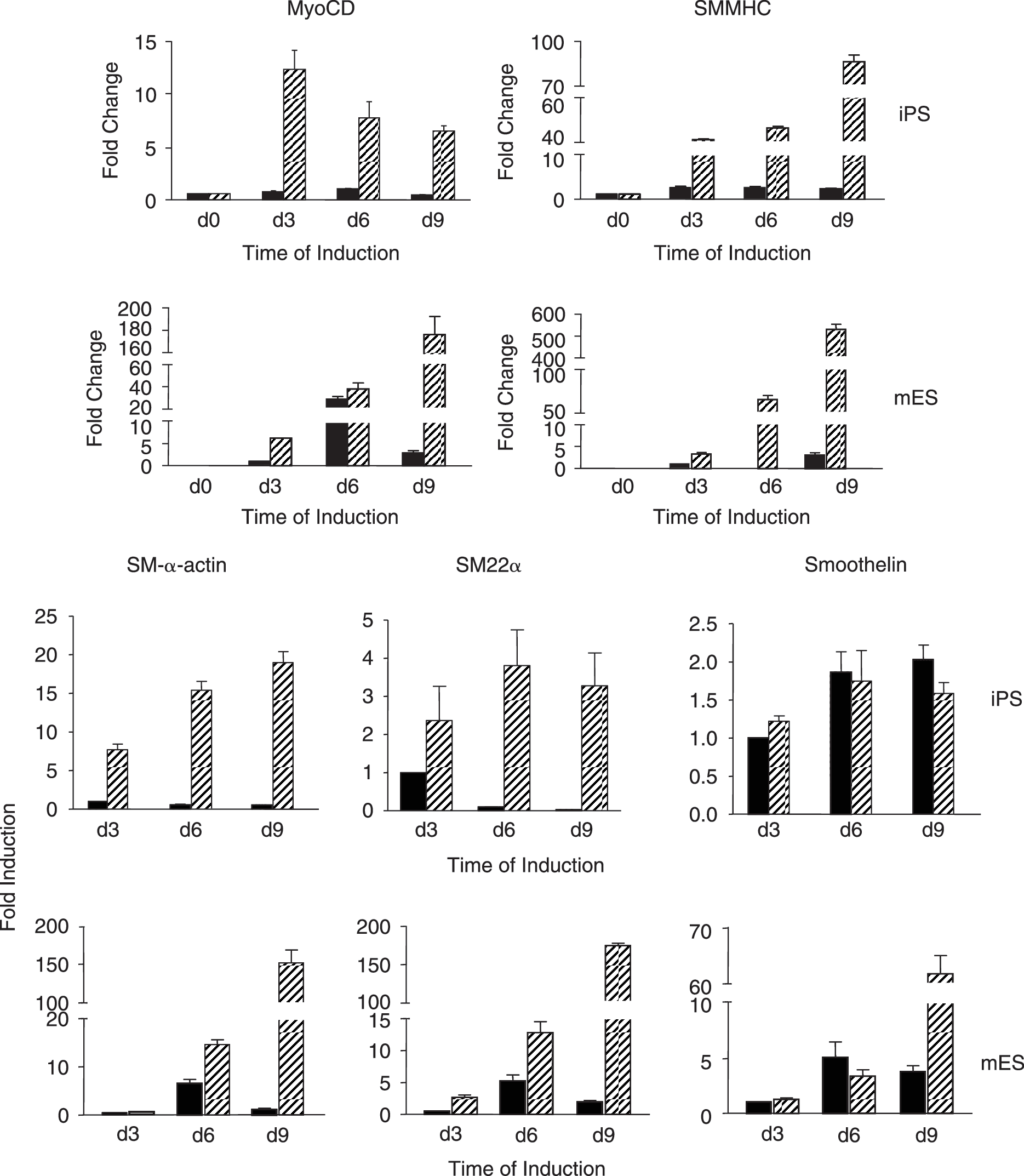
SMC-specific gene expression measured by qRT-PCR analyses in RA-induced differentiation of iPS and mouse ES cells. Total RNA was isolated from DMSO- or RA treatment-derived cells by Day 3, 6, and 9. The expression levels of SMC-specific genes included myocardin, SMMHC, SM22α, SM-α-actin, and smoothelin were determined. Values for mRNA levels normalized by 18S rRNA levels were expressed as mean ± SD (n = 3). Upper panel shows gene expression from O9 iPS cells, lower panel from CMTI-1 ES cells (mouse ES). Solid black bars represent the samples from DMSO-treated cells, hatched bars from RA-treated cells.
Contraction in response to stimuli, which is a requisite characteristic of mature SMCs was checked separately in differentiated cells from mouse ES and O9 iPS cells derived from this protocol. These derived SMCs were treated with carbachol (1 mmol/L), and monitored for 10 min under the microscope (Fig. 3, O9 iPS-derived SMC, mouse ES data not shown). A large proportion (∼80%) of SMC cells derived from RA-treated O9 iPS cells changed their shape in response to carbachol treatment. No obvious response to carbachol was observed on cells derived from DMSO treatment (data not shown). Moreover, SMCs derived from O9 iPS and ES cells exhibited similar functional Ca2+ responses as confirmed by real-time confocal imaging of Ca2+ release upon treatment with the potent vasoconstrictor caffeine (5 mmol/L, Fig. 4), endothelin (3 mmol/L) and the depolarization agent KCl (15 mmol/L) (data not shown).
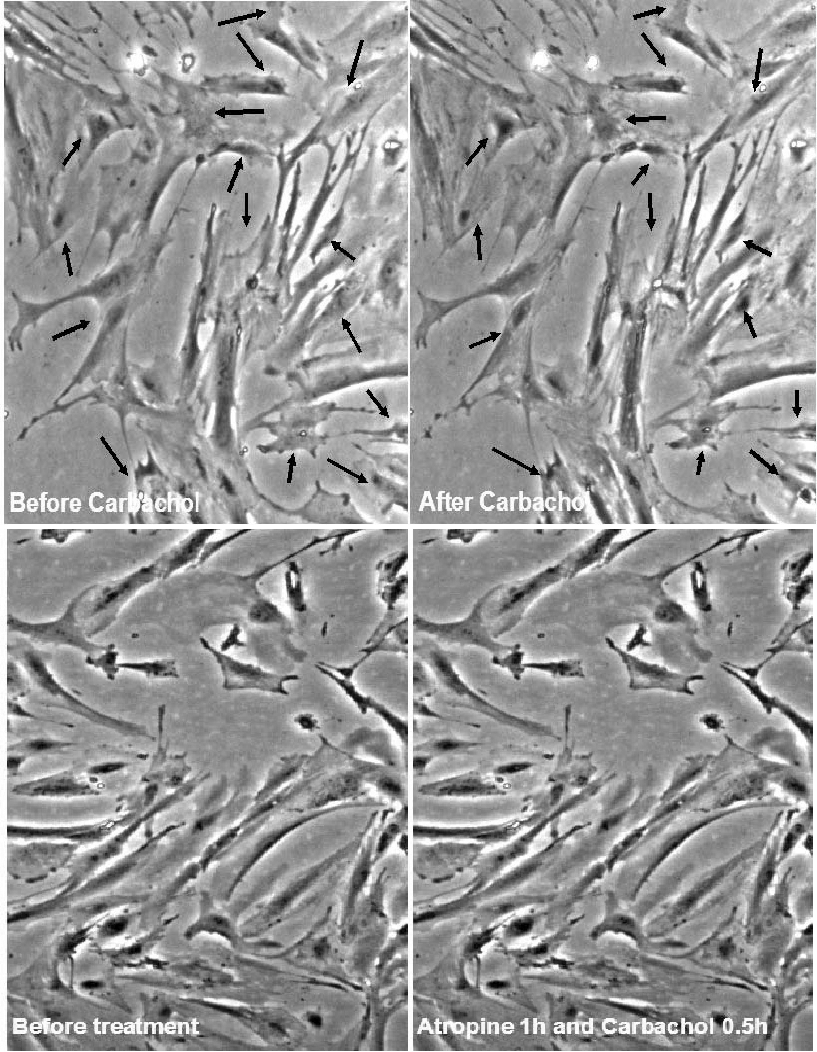
SMC-like cells derived from mouse iPS cells acquired the ability to contract in response to carbachol. SMC-like cells derived from mouse iPS cells after 10−5 mol/L RA treatment were rinsed with PBS and refreshed with culture medium. Upon addition of 1 mmol/L carbachol, pictures were taken at 10 s intervals for 10 min to document the morphological changes under the microscope. Upper panel shows cell contraction before and after carbachol treatment. The arrows indicate the contractile points. Lower panel shows cell contraction in response to carbachol is abolished with pretreatment of 10 μM atropine for 1 h.
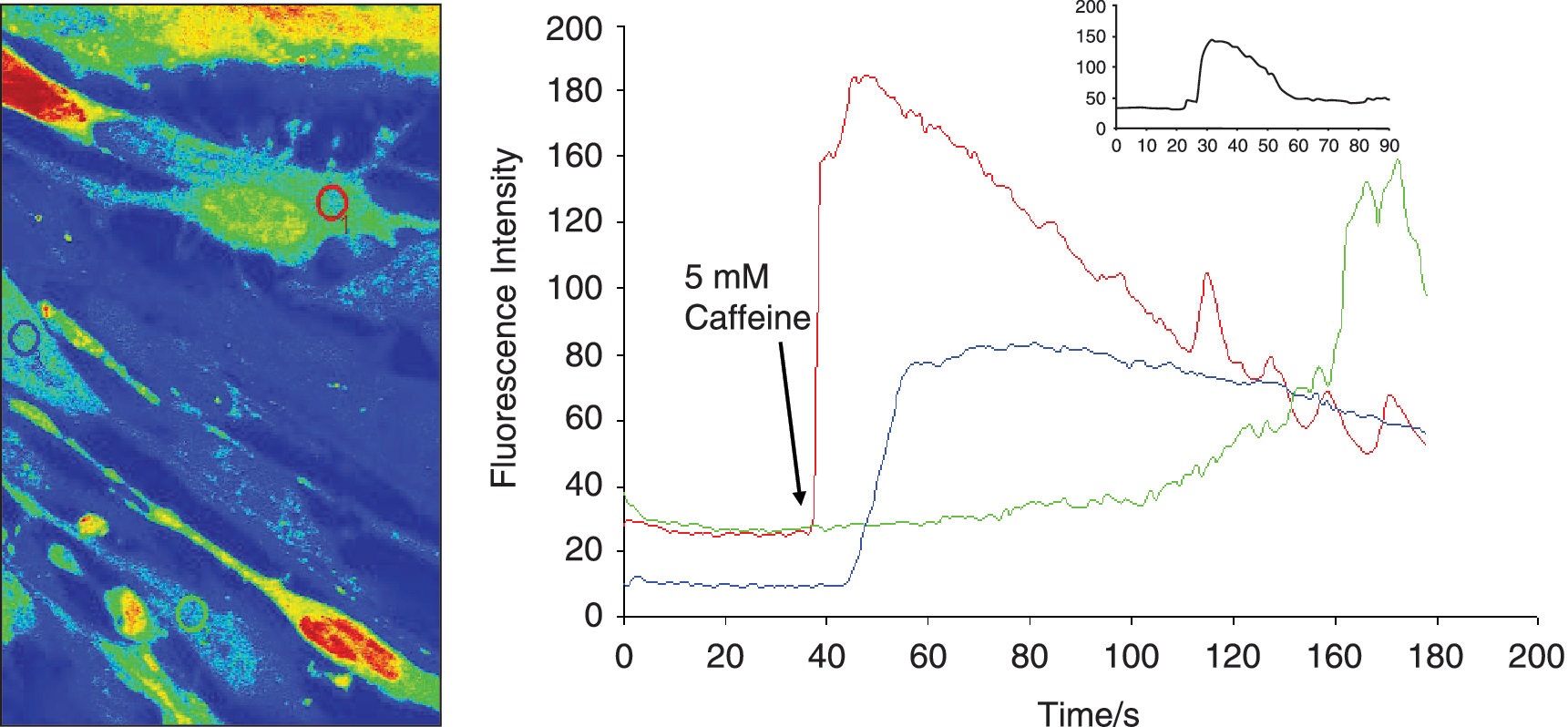
Functional characteristics of SMCs derived from iPS cells. Measurement of calcium transients before and after treatment with the vasoactive agonist (5 mmol/L caffeine). Fluorescence intensity was monitored in cells loaded with Fluo-4 AM with confocal microscope (LSM 510 Meta, Carl Zeiss MicroImaging Inc.). The color lines in the right panel correspond to the fluorescence changes in the cells indicated by the same color circles in the left panel. The inserted graph shows the fluorescence intensity captured from normal mouse SMCs. The arrow shows the time point when caffeine was applied.
Discussion
Mouse ES cells have been used to study SMC differentiation for decades. Previous studies by Drab et al. have shown that treatment of ES-derived embryoid bodies (EBs) with RA and dibutyryl-cyclic adenosine monophosphate could induce differentiation of spontaneously contracting SM-like cell clusters in 67% of EBs [10]. Also, Drab et al. characterized those SM-like cells as vascular SMC based on the expression of specific SMMHC isoforms and typical ion channels as well as the response to specific agonists with increased calcium [10]. Recently, FACS sorted Flk+ cells from mouse iPS-EBs were shown to be able to differentiate into SMC with the treatment of platelet-derived growth factor-BB [15,16]. However, because the lack of defined structural organization and positional information within EBs during pluripotent cell differentiation results in heterogeneity both within and between EBs, this method suffers difficulties in cell-lineage manipulation and process monitoring [17,18].
RA can induce differentiation of pluripotent cells into multiple cell lineages in a dose- and time-dependent manner, including skeletal muscle cell [19], neural cells (10−7 mol/L) [20], SMCs (10−9–10−6 mol/L) [10,21] and myocardial cells (10−9–10−8 mol/L) [22]. Our previous work has also shown that higher concentration of RA (10−5 mol/L) can induce human ES cells to differentiate into SMCs using monolayer culture [12]. Similar to mouse and human ES cells, in the current study, we document that RA (10−5 mol/L) can also induce iPS cells to differentiate into SMCs in vitro with high efficiency. During this process, high concentration RA-treated mouse iPS and ES cells stably acquired SMC characteristics including SMC specific contractile apparatus, transcriptional factor expression and calcium influx response to stimuli. Notably, this platform is EB-independent and does not involve cell sorting. This system is helpful not only to generate patient-specific SMC in vitro, but to monitor the process of SMC differentiation.
Although both mouse ES and iPS cells can differentiate into SMCs in response to RA treatment, we observed differences in the process of differentiation of these two cell lines, as shown in Figs. 1 and 2. For example, the protein levels of SMMHC observed in iPS-derived SMCs after day 8 of RA treatment were lower than the levels observed in ES cell–derived SMC (Fig. 1C). Moreover, the pattern and the levels of expression of myocardin and SMMHC mRNA were different between ES cell–derived and iPS-derived SMC (Fig. 2). Previous studies have also shown that iPS cells possess difference from ES cells [16]. It is possible that these differences are due, at least in part, to an underlying regulation signaling pathway. Further studies are needed to address this question. In addition, TT025 and O9 iPS cells exhibited different responses to high concentration of RA. Although TT025 iPS cells died before day 4, O9 iPS cells were able to differentiate into SMCs. This different reaction may be due in part by the random insertion of those viral vectors of four transcriptional factors used for generation of iPS cells. Moreover, these differences suggest the requirement of comparison of multiple iPS cell lines, when iPS cells will be used for differentiation toward specific cell lineages in the current stage. Regardless of these uncertainties, generation of patient-matched pluripotent stem cells has the potential to address many of the current limitations observed in the treatment of SMC-related diseases. However, before iPS cells can be used as a cell resource for the treatment for cardiovascular disease, c-Myc upregulation during RA-induced differentiation, as observed in Supplementary Fig. S3, has to be paid more attention, given its tumorigenic feature. Several modified protocols for the generation of iPS cells either without requirement of Myc retrovirus or using combination small molecules with Oct4/Klf4 transfection have been recent developed, which will reduce the risk of tumorigenesis [23,24].
The strategy we have used here is simple, reproducible, and efficient in generating SMCs in vitro from iPS and also mouse ES cells. The foresaid data suggest that 10−5 mol/L RA can drive iPS cells to differentiate into SMCs with high efficiency. The reliable and reproducible iPS-SMC system described herein has remarkable potential for directly testing the functional roles of candidate genes implicated in SMC differentiation and function. More importantly, this iPS-SMC system can be applied for individualized SMC generation for vascular tissue engineering, reconstruction, and personalized drug screening, given iPS pluripotency and potential immune privilege.
Footnotes
Acknowledgments
We thank Dr. Rudolf Jaenisch for kindly providing mouse iPS cells and Dr. Minerva T. Garcia-Barrio for her critical reading of the manuscript. Y.E.C. is an established investigator of American Heart Association (0840025N) and was supported by National Institutes of Health (HL68878, HL89544, HL75397, and HL92421). M.Z. was supported by 973 programs (2007CB947800) from the Ministry of Science and Technology of China.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.