
Abstract
Changes in intracellular Ca2+ concentration ([Ca2+]i) play a central role in neuronal differentiation. However, Ca2+ signaling in this process remains poorly understood and it is unknown whether embryonic and adult stem cells share the same signaling pathways. To clarify this issue, neuronal differentiation was analyzed in two cell lines: embryonic P19 carcinoma stem cells (CSCs) and adult murine bone-marrow mesenchymal stem cells (MSC). We studied Ca2+ release from the endoplasmic reticulum via intracellular ryanodine-sensitive (RyR) and IP3-sensitive (IP3R) receptors. We observed that caffeine, a RyR agonist, induced a [Ca2+]i response that increased throughout neuronal differentiation. We also demonstrated a functional coupling between RyRs and L- but not with N-, P-, or Q-type Cav1 Ca2+ channels, both in embryonal CSC and adult MSC. We also found that agonists of L-type channels and of RyRs increase neurogenesis and neuronal differentiation, while antagonists of these channels have the opposite effect. Thus, our data demonstrate that in both cell lines RyRs control internal Ca2+ release following voltage-dependent Ca2+ entry via L-type Ca2+ channels. This study shows that both in embryonal CSC and adult MSC [Ca2+]i is controlled by a common pathway, indicating that coupling of L-type Ca2+ channels and RyRs may be a conserved mechanism necessary for neuronal differentiation.
Introduction
C
Calcium influxes through these intracellular stores may participate in neuronal differentiation, a process in which the role and pathways of Ca2+ signaling have been extensively investigated and modeled [15 –19]. However, it is not clear whether particular Ca2+ pathways are cell-line-specific or if there are common processes in the neuronal differentiation of diverse cells lines. We thus characterized Ca2+ signaling in stem cell lines as distinct as embryonal carcinoma stem cells (CSC) and adult murine bone-marrow mesenchymal stem cells (MSC), to identify conserved events in the control of cell proliferation and differentiation. We have previously shown that P19 embryonic CSC express functional VGCCs, which produce Ca2+-induced Ca2+ release (CICR) through a combination of L-type Ca2+ channels and RyRs and IP3Rs activity [19,20]. We now report that the RyR agonist caffeine produces a transient increase in Ca2+ that becomes larger throughout neuronal differentiation both in CSCs and MSCs. Moreover, these Ca2+ transients are blocked by L-type Ca2+-channel blockers, suggesting that RyRs and L-type Ca2+ channels are coupled. Our data also show that agonists of both of these channels increase neurogenesis and differentiation into neurons in CSCs and MSCs, while antagonists of these channels block these processes. Thus, this report suggests that coupling of L-type Ca2+ channels and RyRs is a mechanism that may be conserved in neuronal differentiation in diverse stem cell lines, and the IP3Rs have a major role in induction of neuronal differentiation beyond the RyRs.
Materials and Methods
Reagents
Unless indicated otherwise, all reagents were purchased from Sigma (Sigma, St. Louis, MO). Appropriate secondary biotin conjugate antibodies were used at 1/200 dilutions (Santa Cruz Biotechnology, Heidelberg, Germany). Primers for real-time PCR were synthesized by Integrated DNA Technologies (Coralville, IA).
Culture and neuronal differentiation of CSC cells
P19 EC cells were grown in DMEM (Invitrogen Corp., Carlsbad, CA) supplemented with 10% of final volume with fetal bovine serum (FBS; Cultilab, Campinas, Brazil), 100 units/mL penicillin, 100 µg/mL streptomycin, 2 mM sodium pyruvate, and 2 mM
Isolation and culture of sphere-derived cells from murine bone marrow
All experiments in this study were performed in accordance with the Animal Protection Guidelines of University of São Paulo. Bone-marrow cells (BMCs) were obtained from five mice for each experiment as previously described [23]. Briefly, bone marrow was collected from 2-month-old C57Bl/6 mice by flushing femurs and tibias with complete medium constituted of Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal calf serum (FCS; Sigma, St. Louis, MO), 2 mM glutamine and 50 U/mL penicillin, 50 mg/mL streptomycin supplemented with heparin at a final concentration of 5 U/mL. Cells were then washed twice in a heparin-free medium and plated in a Petri dish at a density of 2 × 106 cells/cm2. After 2 days, nonadherent cells were removed by two to three washes with phosphate-buffered saline (PBS) and adherent cells were further cultured in complete medium for four supplementary days. To remove differentiated hematopoietic cells, these cells were depleted from whole BMCs by means of immunomagnetic separation with Dynabeads according to the manufacturer’s protocol. Briefly, BMCs were stained with primary rat–anti-mouse antibodies, namely anti-CD4, CD8, CD11b, CD45R/B220, Gr-1, and Ter119 (all purchased from PharMingen, San Jose, CA). After centrifugation of the labeled cells, Dynabeads M-450 Sheep anti-Rat IgG (Dynal Biotech, Oslo, Norway) were added at a concentration of 10 beads per cell. The mixture was placed on a Dynal Magnetic Particle Concentrator (Dynal Biotech) for 1 min and the supernatant containing the line-depleted BMCs was collected. Immunodepleted BMCs were resuspended in 10 mL of complete medium and plated on a 35 × 10 mm tissue culture dish (NUNC, Naperville, IL). Cells were cultured in a humidified incubator containing 5% CO2 and 5% O2 at 37°C. After 24 h, nonadherent cells were removed by washing with PBS and a fresh complete medium was added. Afterwards, the medium was changed twice a week for 3–4 weeks, when cells first reached 70–80% confluency. Then, adherent cells were detached with trypsin/EDTA for 3 min at 37°C. Only easily detached cells were plated on a 35 × 10 mm standard tissue culture dish (first passage). After the first passage, cells were cultured under similar conditions, except that the split ratio was set to 1:2. After additional 5–7 passages, cells were detached at a lower cell density (40–50% confluence). The same conditions were used for subsequent cultures after small round and oval-shaped cells had become predominant by the sixth to eighth passage.
A total of 1 × 105 cells was suspended in complete medium in each well of a six-well ultra low attachment plate (NUNC, Naperville, IL). Cells were cultured without having the medium changed, and were rotated gently every 6 h. The spheres gradually increased in size and, 7 days after suspension, were picked and dissociated mechanically by gentle pipetting. Dissociated cells were cultured in complete medium on standard tissue culture dishes and the medium was changed every 2 days. After reaching 40–50% confluency, cells were detached with trypsin/EDTA and replated at a 1:4 dilution. Sphere-derived cells were harvested for the experiments described below when a single well of the original culture contained >1 × 107 cells.
In order to induce neuronal differentiation, sphere-derived cells were treated as previously described [24] with some modifications. Briefly, cells were plated at a cell density of 1 × 104 cells/cm2 on gelatin-coated dishes in neurobasal medium (Gibco BRL, Grand Island, New York) with B27 Supplement (50×; Invitrogen Corp., Carlsbad, CA), 20 ng/mL EGF, and 10 ng/mL bFGF for 7 days, followed by culture in neurobasal medium with 0.5 µM retinoic acid and 20 ng/mL β-NGF for 5 days.
Immunofluorescence studies
For immunofluorescence, cells were fixed in 1% paraformaldehyde (PFA), permeabilized with 0.2% Triton X-100 in PBS at 4°C for 30 min, blocked with 0.5% casein and 5% normal goat serum in PBS at 4°C for 30 min, and sequentially incubated with anti-nestin (1:200; Chemicon International Inc., Temecula, CA) or NEL (1:100; Santa Cruz Biotechnology Inc., Delaware, CA, USA) antibodies at 4°C overnight, followed by incubation with secondary antibodies coupled with cytochrome 3 (Cy3, 1:200; Jackson Immunoresearch, West Grove, PA) at room temperature (RT) for 1 h. Between each step, cells were washed three times with 0.1% Tween in PBS. Fluorescence was detected and photographed with an AxioCam photomicroscope (Carl Zeiss Vision GmbH, Hallbergmoos, Germany). The immunofluorescent staining of a single sphere was performed as previously described [25].
Confocal [Ca2+]i imaging
[Ca2+]i was measured in single cell as previously described [19,21,22]. The undifferentiated and differentiating cell cultures were incubated with 2.5 µM Ca2+-sensitive dye Fluo3-AM (dissolved in DMSO) plus 0.02% Pluronic F-127 (dissolved in water; Molecular Probes Inc., Eugene, OR) in DMEM medium supplemented with FBS for 30 min at 37°C. The osmolarity of all the solutions ranged between 298 and 303 mosmol/L. Measurements of [Ca2+]i were performed at 37°C in extracellular medium (EM) containing (in mM): 140 NaCl, 3 KCl, 1 MgCl2, 2.5 CaCl2, 10 HEPES, pH 7.4, 10 glucose with the Zeiss Microscope Photometer System (FFP; Carl Zeiss Optronics GmbH, Oberkochen, Germany), based on an inverted microscope (Axiovert 100; Zeiss). Fluo3 fluorescence was excited with a 488 nm line of an argon ion laser, and the emitted light was detected using a bandpass filter at 515–530 nm. Fluorescence values were corrected for background and dark current. Calcium imaging was performed with a LSM 510 confocal microscope (Carl Zeiss Jena GmbH, Jena, Germany), using image sizes of 256 × 256 pixels and acquisition rates of one frame per second. The calibration of the fluorescence signal, in terms of free Ca2+ concentration, was based on the procedure described by Grynkiewicz and collaborators [26]. At the end of each experiment, 5 µM ionophore (4-Br-A23187) followed by 10 mM EGTA were used to determine F max and F min fluorescence values, respectively. The average baseline fluorescence was normalized to F min as previously described [19,21]. To evaluate the contribution of VGCC activation to the studied Ca2+ signals, the depolarizing stimuli were delivered in the presence of the nonselective VGCC blocker Cd2+ (250 µM). Before testing the effects of Cd2+, the stability of [Ca2+]i responses to depolarizing stimuli repeated at 5-min intervals was evaluated. The amplitude of the ΔF/F ratios in response to the first four stimulations remained fairly stable; those of the third and fourth transients were, respectively, 4.9 ± 2.3 % (n = 45) and 8.5 ± 2.1 % (n = 37) smaller than that of the first.
The role of Cav1-mediated Ca2+ influx in promoting neuronal differentiation of neuronal progenitor cells (NPC) derived from CSC and MSC was also investigated. Depolarization-induced Ca2+ transients were studied in cells cultured in defined medium with the addition of either 5 µ
For calcium imaging in Ca2+-free extracellular medium, cells were treated for 1 min with Ca2+-free medium containing 1 mM EGTA. Control measurements in the presence of extracellular Ca2+-containing medium were performed before and after experiments in Ca2+-free medium. All antagonists were preincubated with CSCs or MSCs for, at least, 20 min before experiments were performed.
Western blot assays
NPC from day 6 derived from CSC and MSC were homogenized in ice-cold lysis buffer containing protease inhibitors, 10 mM Tris-HCl (pH 7.5), 8 M urea, 20 mM EDTA, 150 mM NaCl, 1% Triton X-100. Forty micrograms of membrane proteins that were fractionated by differential centrifugation, separated by SDS-PAGE, and transferred to nitrocellulose membranes were used. The procedure of incubation with primary and secondary antibodies was essentially the same as described previously [18,21]. The following polyclonal rabbit antisera were used: anti-nestin (1:200), anti-neuronal enolase (NEL, 1:100), anti-SSEA-1 (1:100; Santa Cruz Biotechnology, Heidelberg, Germany), and anti-β-actin (1:400). β-actin protein expression was used as internal standard for relative quantification of nestin and NEL expression levels.
RNA isolation, reverse transcription, real-time PCR, and conventional PCR
Total RNA was isolated using TRIzol (Invitrogen Corp., Carlsbad, CA) from undifferentiated cells and from days 2 to 8, following the induction of neuronal differentiation in the presence of RA for CSC and in the presence of RA and β-NGF for MSC. Additional RNA was extracted on the sixth day of neuronal differentiation that had been cultured for 48 h in the presence or absence of 5 µM nifedipine or 10 µM Bay-K. Integrity of the isolated RNA was verified by separation of an aliquot of the extracted RNA on a 2% ethidium bromide-stained agarose gel. DNA was removed from RNA samples by incubation with DNase I (Ambion Inc., Austin, TX).
Three micrograms of total RNA were reverse transcribed to cDNA with 200U of RevertAid H Minus M-MuLV-reverse transcriptase (Fermentas Inc., Hanover, MD). DNA templates were amplified by real-time PCR on the 7000 Sequence Detection System (ABI Prism; Applied Biosystems, Foster City, CA) using the Sybr green method [19,22] or were amplified by PCR and analyzed as described previously [18,19,22]. Variations in cDNA concentrations were normalized with β-actin as an internal control, which is a constitutively expressed gene. Experiments were performed in triplicate for each data point. Primer sequences for reverse transcription and quantitative PCR amplification (qRT-PCR) of β-actin, IP3Rs and RyRs isoforms mRNA and for PCR amplification (RT-PCR) of β-actin, nestin, and NEL cDNA used in this study are listed in Table 1. Negative controls were conducted on water and on total RNA nonreverse transcribed. To evaluate the specificity of the RT-PCR products, they were gel-purified, cloned into PGEM T-Easy vectors (Promega, Madison, WI), sequenced, and confirmed to be identical to mouse NEL and nestin cDNA.
Proliferation assays
5-Bromo-2-deoxyuridine (BrdU; Amersham Life Sciences, Arlington, IL, EUA), a pyrimidine analog, was used to determine the synthesis rate of DNA and to perform an incorporation assay. Twenty-four hours prior to proliferation stimulation, NPC were kept in serum-free medium. They were then maintained for 8 h in culture medium in the presence or absence of 1 µM ryanodine (Rya ExC, acts as an activator of ryanodine-sensitive calcium stores) or 10 µM ryanodine (Rya InhC, an inhibitor of ryanodine-sensitive calcium stores), which were co-applied with or without agonists or inhibitors of receptor-induced calcium signal transduction. BrdU (15 µM) was then added to the cell culture and kept for an additional hour. Cell proliferation was detected by using an anti-BrdU monoclonal antibody (Roche Applied Science, Indianapolis, IN), biotinylated goat anti-mouse IgG, and avidin–biotin complex (Jackson Immuno Research, West Grove, PA; 1:200). Aminoethylcarbazole (AEC) was used as a chromogen for visualization of immunostained cells. Further details are provided elsewhere [18,21].
The effects of Rya ExC, Rya InhC, 10 µM Bay-K, an agonist of L-type Ca2+ channels, 5 µM nifedipine, a blocker of VGCCs, 10 µM cyclopiazonic acid (CPA), a sarcoplasmatic/endoplasmatic reticulum Ca2+-ATPase (SERCA) inhibitor, 10 µM xestopongin C (XeC), a membrane-permeable inhibitor of IP3 and of SERCA pumps [27,28], adenophostin-A as agonist for IP3Rs [29,30], and 20mM KCl as a depolarizing agent were evaluated for proliferation and neuronal differentiation induction on NPC.
In immunofluorescence double-labeling experiments, NPC were fixed in acid alcohol and processed for NEL or nestin staining, followed by BrdU staining. Cell preparations were then incubated with rabbit anti-NEL or anti-nestin antibodies (1:100; Santa Cruz Biotechnology, Delaware, CA), and immunofluorescence was detected in the presence of anti-rabbit IgG-Cy3 (Abcam, Cambridge, MA) or IgG-Alexa-Fluor 488 (Molecular Probes Inc., Eugene, OR) secondary antibodies. Incubations were performed for 1 h at room temperature. NEL or nestin immunostaining was followed by incubation of the cell preparation with 1 N HCl, neutralization with 0.1 M sodium tetraborate, and incubation with Alexa Fluor 647-conjugated anti-BrdU (Molecular Probes Inc., Eugene, OR) monoclonal antibody for 1 h at room temperature. After washing with PBS, BrdU/NEL, or BrdU/nestin, stained cells were examined by fluorescence microscopy [18,21].
Statistical analysis
Data are represented as means ± SEM of five or more independent experiments (each with two replicates). The peak amplitude of [Ca2+]i increase/response is represented in the bar diagrams with mean ± SEM. Statistical significance was determined by Student’s t-test or one-way ANOVA plus a post-hoc Tukey’s test. Values of P < 0.05 were considered as statistically significant.
Results
Resting [Ca2+]i levels
Basal [Ca2+]i levels were 31 ± 3.9 nM in embryonic CSCs (n = 101) and 72 ± 8.4 nM in the eighth day of neuronal-differentiated CSCs (n = 98). For undifferentiated MSCs, basal [Ca2+]i levels were 47 ± 5.7 nM (n = 86) and 102 ± 12.6 nM after neuronal phenotype acquisition (n = 74). Under resting conditions, the basal (resting) [Ca2+]i levels in these differentiating cells varied, depending on the day of culture, from 51 ± 5.0 nM (CSC, n = 48) and 62 ± 6.7 nM (MSCs, n = 57) on the fourth day after RA or RA and β-NGF addition, respectively, to 74 ± 9.8 nM (n = 64) and 87 ± 10.1 nM (n = 72) on the sixth day. Although small variations of the resting [Ca2+]i levels were observed, no clear correlation could be found with the number of days cells were in culture. Data pooled from days 0 to 8 were scattered around a mean value of 57 ± 6.8 nM (CSC, n = 52) and 74 ± 8.8 nM (MSC, n = 61).
Gene expression of RyR and IP3R isoforms during neuronal differentiation
We next studied gene expression of IP3R and RyR isoforms during neuronal differentiation of CSCs and MSCs. At 8 days in vitro, among the three RyR isoforms, only RyR3 and RyR2 were detected in significant levels, in CSCs and MSCs, respectively. All three IP3R isoforms were detected, although IP3R type 1 and 2 mRNA expressions were very weak in CSCs and MSCs, respectively (Fig. 1). Real-time PCR revealed that gene expression of IP3R 1, 2, and 3 subtypes was already present in embryonic CSCs and adult MSCs, remaining at a lower level during initial and intermediate differentiation states and increasing significantly when differentiating cells became post-mitotic and underwent final maturation on days 6–8 (Fig. 1).
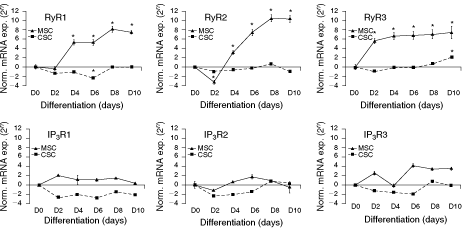
Quantification of the different isoforms of ryanodine (RyR) and inositol-1,4,5-triphosphate (IP3R) receptor mRNA transcription during neuronal differentiation of carcinoma (CSC) and mesenchymal (MSC) stem cells by real-time PCR. Gene expression levels of receptor subtypes were normalized to the internal control β-actin. The graphs show RyR and IP3 mRNA transcription levels in differentiating cells compared to those in undifferentiated cells which were normalized to zero. Cells on day 8 of differentiation were pretreated on day 6 with cytosine arabinoside, in order to avoid contamination of the cell cultures with glial cells. The data shown are mean values ± SEM of at least five independent experiments in triplicate. In some cases, the error bar is included within the symbol. *P < 0.05 compared with control data.
[Ca2+]i responses induced by caffeine increase during neuronal differentiation
Caffeine (10 mM), which in millimolar concentrations acts as an inhibitor of intracellular receptors for IP3, as well as an activator of RyRs [31], was applied for 10 s on undifferentiated and differentiated cells from the fourth day (when the majority of the cells express neuronal progenitor markers), sixth day (start of neuronal maturation), and eighth day (when mature neurons are the main population) after induction of neuronal differentiation [22,32]. As we have previously shown, caffeine did not induce a [Ca2+]i response in embryonic CSCs [19]. Caffeine-induced [Ca2+]i responses in differentiating cells were 381 ± 7.0 nM on the fourth day and 712 ± 17.9 nM on the sixth day after RA induction and increased to 945 ± 34.8 nM after the eighth day. MSC [Ca2+]i responses induced by caffeine were, respectively, 304 ± 13.5 nM, 566 ± 57.2 nM, and 818 ± 12.0 nM on days 4, 6, and 8 after induction of differentiation. Caffeine-induced [Ca2+]i responses during differentiation presented an increase in the [Ca2+]i from embryonic to neuronal mature cells from the eighth day in culture, when the maximum response was observed (Fig. 2).
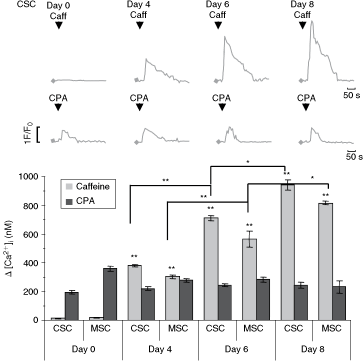
Patterns of caffeine and cyclopiazonic CPA-induced [Ca2+]i increases during neuronal differentiation of carcinoma (CSC) and mesenchymal (MSC) stem cells, respectively. Upper panel: Representative traces of [Ca2+]i responses of CSC cells after application of caffeine (Caff, 10 mM) and then with the SERCA inhibitor cyclopiazonic acid (CPA, 10 µM). Lower panels indicate values of Δ[Ca2+]i increases obtained in the presence of caffeine represented with gray bars or CPA represented with black bars. Similar observations were obtained in five different experiments for each cell line. The total number of cells analyzed in each experiment was 51–63. Data are presented as mean values ± SEM; *P < 0.05, **P < 0.001 compared with control data.
Intracellular calcium stores activated by CPA
IP3Rs activation induces Ca2+ release from intracellular calcium stores in embryonic and neuronal cells [28,33 –36]. In order to check the involvement of Ca2+ release from IP3-sensitive stores, we have tested the [Ca2+]i response induced by 10 µM of CPA applied during 30 s at different days in vitro. CPA (10 µM) was applied for 30 s on cell cultures on days 0, 4, 6, and 8 of differentiation derived from both CSCs and MSCs. First, the CPA-induced [Ca2+]i response was nearly identical when applied twice after a long wash between applications to CSC or MSC. However, these [Ca2+]iresponses had constant Δ[Ca2+]i values throughout the differentiation process, differently from the caffeine-induced calcium transients, which increased progressively across days (Fig. 2).
Spontaneous [Ca2+]i oscillations were observed under resting conditions or after a brief application of caffeine throughout the differentiation. Neuronal-differentiated (eighth day) cells showed a mean [Ca2+]i peak amplitude of about 2.6 ± 0.03 for CSC- and 3.6 ± 0.05 for MSC-derived neurons (DN). These responses were diminished when cells were incubated with 5 µM nifedipine (N- or P/Q-type Ca2+-channel blockers had no effect—data not shown) or 1 µM thapsigargin (inhibitor of intracellular calcium stores), recovering to normal levels after washout of these drugs (data not shown).
P
FWD, forward primer; REV, reverse primer.
Caffeine- and CPA-specificity action
Caffeine-induced [Ca2+]i responses were initially tested in the presence of 10 µM ryanodine to verify the specificity of caffeine action on the [Ca2+]i increases. Ryanodine at a concentration of 10 µM (ryanodine inhibitory, or Rya InhC) acts as an inhibitor of RyRs, while 1 µM ryanodine (ryanodine excitatory, or Rya ExC) excites RyRs. Neuronal-differentiated cells from the eighth day were used for this test. In control conditions, caffeine-induced [Ca2+]i responses were (in nM): 1008 ± 50.4 and 957 ± 56.0 for CSC- and MSC-DN, respectively (data not shown). To avoid any additional [Ca2+]i elevations ryanodine alone was applied for 2 min, inducing a very small increase in [Ca2+]i to 105 ± 15.7 nM (CSC-DN) and 123 ± 10.2 nM (MSC-DN). After preincubation with RyA InhC, caffeine induced a very small [Ca2+]i elevation in neurons derived from both, CSCs (Δ[Ca2+]i = 47 ± 6.1 nM) and MSCs (Δ[Ca2+]i = 164 ± 17.8 nM). No significant inhibition of [Ca2+]i increases was obtained with the IP3R inhibitor XeC (10 µM) on both, CSC and MSC. These results indicated that Rya InhC inhibited caffeine-induced [Ca2+]i responses.
In order to evaluate the specificity of CPA activity on intracellular calcium stores, XeC (10 µM) was used to inhibit IP3Rs and SERCA pumps (data not shown). CPA-induced [Ca2+]i responses were of 242 ± 38.6 nM and 254 ± 27.2 nM on CSC-DN and MSC-DN, respectively, and these responses were practically absent when the culture cells were preincubated with XeC for 5 min (Δ[Ca2+]i in nM: 17 ± 2.2 and 35 ± 10.7, respectively). The inhibition of the IP3Rs and the SERCA pumps responses caused by the presence of XeC was reversible as receptor responses recovered following washing out of the inhibitor (Δ [Ca2+]i in nM: 234 ± 35 for CSC-DN and 242 ± 29 for MSC-DN).
P
Rya InhC, treatment with ryanodine at inhibitory concentration (10 µM); Rya ExC, treatment with ryanodine at excitatory concentration (1 µM); AdA, Adenophostin-A (2 nM); xestopongin C (XeC, 10 µM); Nifedipine was used at 5 µM, Bay-K at 10 µM, and KCl at 20 mM; (—) data not evaluated.
Data presented are means ± SEM of positive-stained NPC for the neuronal enolase (a mature neuronal marker, NEL), nestin (a marker for NPC) and BrdU/NEL and BrdU/nestin double-labeling. *P < 0.05, **P < 0.001.
[Ca2+]i responses induced by Cav1 increase during neuronal differentiation
To further characterize L-type Cav1 channel and RyR [Ca2+]i responses, 10 µM Bay-K, an agonist of L-type channels, or 1 µM ryanodine (Rya ExC) were applied. Each drug was applied for 30 s on CSC and MSC cells cultures during neuronal differentiation. L-type Cav1 channel-induced [Ca2+]i responses increased during the differentiation process for both, CSCs and MSCs. The Δ[Ca2+]i in nM for embryonic CSCs was 148 ± 12.4, and for differentiating cells it was 303 ± 15.9, 694 ± 36.2, and 970 ± 36.8 at 4, 6, and 8 days after induction with RA. The response for undifferentiated MSCs was 226 ± 14.7, while it was 419 ± 11.1, 693 ± 18.2, and 885 ± 39.1 at days 4, 6, and 8 following induction with RA and β-NGF (Fig. 3A). As demonstrated for Bay-K application and confirming real-time PCR and caffeine-induced [Ca2+]i transients data, ryanodine-induced [Ca2+]i responses increased during the differentiation of CSC and MSC (Δ[Ca2+]i in nM for: embryonic CSC, 17.9 ± 4.9; fourth day, 290 ± 7.0; sixth day, 723 ± 40.3; and 857 ± 34.8 at eighth day. And for MSC: undifferentiated MSC, 15 ± 1.8; fourth day, 290 ± 6.9; sixth day, 771 ± 40.6; and 939 ± 13.0 at eighth day; Fig. 3A).

Functional characterization of L-type Cav1 channel and RyR [Ca2+]i responses. (
To elucidate if RyR or L-type Cav1 channel activation controls [Ca2+]i transients of each other, VGCCs were activated with 20 mM KCl and these responses were significantly inhibited by ryanodine InhC (Fig. 3B). Furthermore, [Ca2+]i transients mediated by 10 µM Bay-K were also significantly inhibited by 10 µM ryanodine (Fig. 3B). These results indicated that L-type Cav1 channel-induced [Ca2+]i responses were mainly controlled by RyRs in neuronal-differentiated cells derived from CSC and MSC, with a minor role played by N-type channel for the latter.
L-type Cav1 channels are controlled by caffeine-induced [Ca2+]i responses
The role of the various VGCCs expressed by neuronal-differentiated CSCs and MSCs cells on caffeine-induced [Ca2+]i responses was studied in order to evaluate the contribution of extracellular Ca2+ to neuronal differentiation. Blockers of VGCCs, such as nifedipine (for L-type Cav1 channel), ω-CgTx (for N-type), and ω-MVIIC (for P/Q-type Cav2 channels), were used to evaluate the participation of these channels in caffeine-evoked [Ca2+]i increases (data not shown). These VGCC blockers were preincubated with the cells for 15 min at room temperature in the dark prior to application of caffeine. The first application of caffeine evoked an increase of 1016 ± 55.6 nM and 892 ± 45.0 nM for CSC-DN and MSC-DN, respectively. Caffeine-induced [Ca2+]i responses were strongly reduced in the presence of 5 µM nifedipine in both, CSC-DNs and MSC-DNs (Δ[Ca2+]i in nM: 246 ± 21.4 and 343 ± 15, respectively). The N-type channel blocker, ω-CgTx-GVIA (800 nM), and the P-type channel blocker, ω-MVIIC (100 nM), presented no effects on caffeine-induced [Ca2+]i responses in CSC-DNs (in the presence of ω-CgTx-GVIA, Δ[Ca2+]i = 1030 ± 46.6 nM; and in the presence of ω-MVIIC, Δ[Ca2+]i = 987 ± 49.7 nM, relative to control conditions Δ[Ca2+]i = 951 ± 62.7 nM). However, for MSC-DNs the N-type channel blocker promoted a significant reduction on [Ca2+]i elevations by caffeine (Δ[Ca2+]i = 674 ± 111.7 nM, relative to control conditions Δ[Ca2+]i = 862 ± 36.6 nM).
Role of L-type Ca2+ channels in inducing NPC cells differentiation
In order to investigate the role of Cav1-mediated Ca2+ influx in promoting neuronal differentiation of NPCs, cells from the fourth day of differentiation were cultured for 48 h in the presence of an antagonist (5 µ
The percentage of NEL-positive stained cells treated with nifedipine dropped 62% and 54% relative to control conditions for cells derived from CSC and MSC, respectively (Fig. 4A and Table 2). NEL-positive cells in Bay-K-treated cultures increased 36% and 23% relative to control for CSC- and MSC-DN, respectively. Similar effects were observed when NPC derived from CSC were stained for nestin (23% ± 4.1% of the cells cultured with nifedipine, 32% ± 3.0% of control cells, and 41% ± 3.4% of those cultured with Bay-K) and from MSC (23% ± 3.0% of the cells cultured with nifedipine, 31% ± 2.8% of control cells, and 38% ± 4.0% of those cultured with Bay-K) (Fig. 4A and Table 2). In agreement, nestin and NEL gene and protein expression were enhanced as revealed by RT-PCR and Western-blot experiments when NPC of CSC or MSC had been differentiated in the presence of either DHPs (Fig. 4B,C, see Table 2 for summary). These results suggest that Cav1 channels also participate in induction of differentiation of NPC.
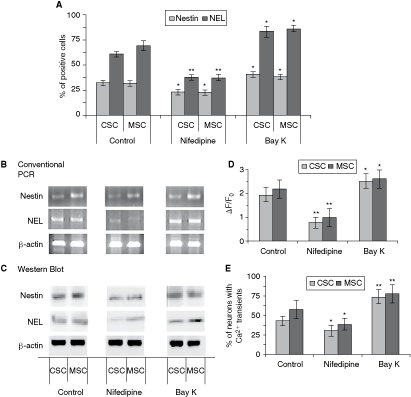
Cav1-mediated Ca2+ influx induces neuronal progenitor cells (NPCs) to differentiate into neuronal cells. NPCs from day 4 of differentiation derived from carcinoma (CSC) and mesenchymal (MSC) stem cells were cultured for 2 days in the presence or absence of nifedipine (5 µ
To quantify cell proliferation, NPC on the fourth day were used in a BrdU assay. No significant differences were observed on BrdU incorporation between nontreated cells from CSC- and MSC-DN, even when cells were exposed to nifedipine or Bay-K (Table 2). Based on our analysis of 20–23 randomly selected 20 fields for each experimental condition, the total number of cells in control, nifedipine-, and Bay-K-treated cultures were not significantly different. These findings suggest that NPC proliferation in CSC and MSC are not significantly affected by either of the DHPs, suggesting a common role for the CSC and MSC differentiation and proliferation.
In Ca2+-imaging studies performed on neuronal cells from the sixth day of differentiation, KCl stimulation caused very weak increases in fluorescence (ΔF/F = 0.8 ± 0.23 and 1.0 ± 0.42, for CSC- and MSC-DN, respectively) when these cells were cultured with nifedipine, whereas those cells cultured in control conditions displayed signals with a mean amplitude of 1.9 ± 0.29 and 2.2 ± 0.37, for CSC- and MSC-DN, respectively. These were enhanced in the presence of Bay-K (2.5 ± 0.31 and 2.6 ± 0.42, i.e., 29% and 20% larger than in CSC- and MSC-DN controls, respectively) (Fig. 4D). The percentage of nifedipine-treated NPC cells responding to KCl stimulation was also significantly reduced 31% ± 4.8% and 38% ± 7.5%, respectively. In contrast, in NPC cultured in the presence of Bay-K, [Ca2+]i transients were observed in 74% ± 13.4% and 78% ± 10.7% versus 43% ± 6.4% and 57% ± 9.8% of CSC- and MSC-DN controls, respectively (Fig. 4E). No significant effect on cell viability evaluated by Tunel was observed on treated cells (data not shown), supporting the idea that a functional neuronal phenotype may be maintained via activation of L-type Cav1 channels.
RyR direct neural differentiation process to neuronal cell fate
To evaluate if the increased neuronal differentiation in NPC derived from CSC and MSC is directly related to RyR function, neuronal differentiation was analyzed by adding 1 µM (Rya ExC, which excites RyRs) or 10 µM ryanodine (Rya InhC, an inhibitor of RyRs). Treatment of cells in the fourth day of differentiation from CSC and MSC during 48 h with ryanodine ExC resulted in a significant increase in the proportion of NEL-positive cells while ryanodine InhC decreased this percentage to a level significantly smaller than that found in control conditions (see Table 2 for a summary). Likewise, the percentage of nestin-positive cells increased and decreased with ryanodine ExC or InhC treatment, respectively (Table 2).
In order to further determine if the reduced number of NEL-positive cells in ryanodine InhC-treated cultures is due to the inhibition of neuronal differentiation or/and decreased cell viability, double-immunelabeling for BrdU and NEL or BrdU and nestin were performed for both, ryanodine ExC and InhC concentrations. BrdU would mark the proliferative cells that are neural progenitor cells (nestin-positive) or immature neuron cells (NEL-positive). It was found that ExC treatment promoted an increase on BrdU/NEL-positive cells for both, CSC- and MSC-DN, compared to control and decreased on InhC treatment. Likewise, the percentage of BrdU/nestin-positive cells treated with ExC and InhC was significantly increased and decreased relative to control conditions for CSC- and MSC-DN, respectively (Table 2 and Fig. 5A,B). No significant increases on cell apoptosis were noted for both, ExC or InhC treatment on BrdU/NEL- or BrdU/nestin-positive cells (data not shown). These data suggest that RyR-mediated [Ca2+]i elevations induce differentiation of NPCs, regulates the proliferation of immature neurons, and increases the proliferation rate of NPCs.
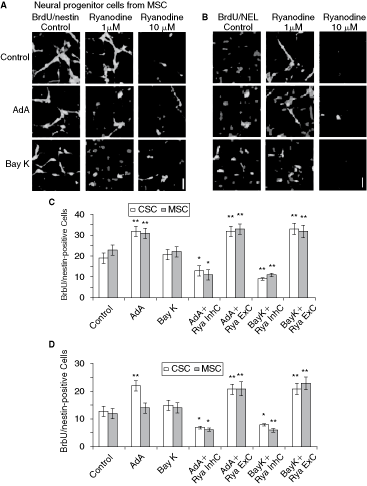
Activation of ryanodine receptors (RyR) promotes neuronal differentiation and proliferation. (
Furthermore the role of IP3R on neuronal differentiation and proliferation of NPC from CSC and MSC was evaluated as well as if there was any correlation with RyR activation or inhibition. The activity of adenophostin-A (2 nM), an IP3R agonist, in NPCs, the presence or absence of ryanodine ExC and InhC were tested. Adenophostin-A and ryanodine ExC promoted an increase of BrdU/NEL-positive cells compared to control without any drug, but with similar results when treated with adenophostin-A alone (Fig. 5B and Table 2). A significant decrease in the percentage of BrdU/NEL-positive cells in ryanodine InhC treatment was observed, and co-treatment with ryanodine InhC and adenophostin-A did not alter this result. This suggests that IP3R participates only on differentiation but not on proliferation. The percentage of BrdU/nestin-positive cells treated with adenophostin-A alone presented an increase on NPC of CSC and MSC relative to control. However, treating NPCs with adenophostin-A and ryanodine ExC or InhC induced a significant increase and decrease, respectively, in cells responses (Table 2 and Fig. 5A). These data suggest that adenophostin-A-mediated [Ca2+]i elevations induce neuronal differentiation of NPC via a pathway independent of RyR and have effects on the proliferation of immature neurons and NPC.
RyR guided the L-type Cav1 channel induction neuronal differentiation
RyRs participate in the control of Ca2+ release through channels in the ER and contribute to the dynamics of Ca2+ signaling in neurons. However, it is not clear whether RyR- or L-type Cav1 channels-induced [Ca2+]i responses contribute to Ca2+ signaling during early neuronal development and if neurogenesis requires Ca2+ signaling by RyR or L-type Cav1 channels. We therefore directly analyzed the functional relationship of RyR and L-type Cav1 channels by applying Bay-K 10 µM, a L-type Cav1 agonist, alone and in the presence of ryanodine ExC or InhC (Fig. 5A,B, and Table 2 for a summary). We found that in NPC from both, CSC and MSC cells, Bay-K alone significantly increased the percentage of NEL- and nestin-positive cells, an effect that was inhibited by the co-treatment with ryanodine InhC. The double-immunelabeling for BrdU/NEL-positive and BrdU/nestin only increased or decreased when NPC were co-treated with Bay-K and ryanodine ExC or InhC, respectively. Similar results were obtained with 10 µM nifedipine treatment (Fig. 6A and Table 2), suggesting that tonic activity of L-type Cav1 channels is important for the neuronal differentiation in NPC but not for its proliferation. On the other hand, neither Bay-K nor nifedipine had any effects on the neuronal differentiation in undifferentiated CSCs and MSCs (Fig. 6B and Table 2). The percentage of NEL-positive cells in CSC and MSC cultures in the presence of Bay-K (1.0% ± 0.5% and 0.9% ± 0.3%) or nifedipine (0.8% ± 0.4% and 0.7% ± 0.4%) were not significantly different from control conditions (1.1% ± 0.6% and 0.8% ± 0.4%). These results demonstrate that the activation of L-type Ca2+ channel promotes neuronal differentiation through its functional coupling with RyRs, and that RyRs affect proliferation of immature neurons and NPCs.

Effects of IP3Rs and L-type Ca2+-channels inhibition on ryanodine receptors activation. (
It has been demonstrated that RyRs can be activated by mobilization of extracellular Ca2+ entry through membrane Ca2+ channels in neurons [37], so we evaluated the role of Ca2+ influx in mediating the neuronal differentiation of NPC from both, CSCs and MSCs, and characterized the functional coupling between RyRs and the regulatory effects of Ca2+ influx on neuronal differentiation. We studied the impact of activation of VGCCs on neurogenesis in cultures treated with ryanodine ExC or InhC in the presence of depolarizing extracellular medium by adding KCl (20 mM). The proportion of NEL-positive cells was increased in conditions with depolarizing medium but decreased when it was co-treated with ryanodine InhC or nifedipine (5 µM) (Table 2). The co-treatment with KCl and nifedipine in the presence of ryanodine ExC or ryanodine InhC led to a significant increase and decrease, respectively, on the percentage NEL-positive cells. These data indicate that RyRs play a common role in neurogenesis in different cell lines, such as CSCs and MSCs, by increasing [Ca2+]i triggered by activation of L-type Ca2+ channels.
Discussion
Spontaneous [Ca2+]i oscillations constitute a hallmark of developmental networks. They have been observed in several neural structures including the retina [38], neocortex [39], hippocampus [40], and spinal cord [41]. Although this spontaneous activity plays a key role in the control of neuronal differentiation and synaptogenesis, very little is known about the cellular and molecular events underlying these phenomena during neuronal differentiation.
We further studied the role of [Ca2+]i oscillations in differentiation using embryonal CSCs and adult MSCs. Initially we demonstrated expression of RyRs in early embryonal carcinoma and mesenchymal adult stem cells for the first time. These receptors and the VGCCs are likely responsible for the calcium-induced calcium release (CICR) controlling the spontaneous [Ca2+]i events. The present data show a transient CICR mechanism in both CSCs and MSCs, involving ER receptors (IP3Rs and RyRs) and plasma membrane L-type channels. The presence of classic RyRs was detected by the caffeine-induced [Ca2+]i increases and its blockage by Rya InhC (10 µM ryanodine). Furthermore, the caffeine-induced [Ca2+]i responses were abolished by the L-type channel blocker nifedipine. While N- (ω-CgTx) and P/Q-type (ω-MVIIC) blockers presented no effects on CSC, a small effect was promoted by the N-type blocker on MSC. It is worth mentioning that nifedipine does not pass through the plasma membrane, so it cannot interact directly with RyRs [42]. Thus, our data reveal a functional coupling between Cav1 L-type channels and RyRs in neurons [37,43], but not in undifferentiated CSC and MSC.
The involvement of IP3Rs in the mobilization of calcium from intracellular stores was also evaluated. The results demonstrate that XeC completely abolished the [Ca2+]i increase induced by the SERCA pump inhibitor CPA, thus indicating that functional IP3Rs are present in undifferentiated CSC and MSC and during their neuronal differentiation. These data led us to conclude that IP3Rs are responsible for early [Ca2+]i elevations in those stem cells. In later stages of the differentiation process, regulation of [Ca2+]i relied on both IP3- and ryanodine-sensitive stores.
Real-time PCR analysis revealed that gene expression of RyRs and IP3Rs was already present in CSC and MSC in a lower level with prevalence of the isoforms RyR2 and RyR3, in accordance with a previous study [44]. While RyR1 and RyR2 function as Ca2+-release channels, it has been recently demonstrated that RyR3 may negatively regulate RyR2 activity [45]. In undifferentiated CSC and MSC, before induction of neuronal differentiation, caffeine induced no [Ca2+]i responses coinciding with the low level of expression of the RyR isoforms. It has been suggested that functional interactions between the three isoforms in stem cells may not allow caffeine-induced [Ca2+]i responses, and that RyR2 expression indicates a mature functional phenotype as shown in various brain regions [34].
In vivo experiments studying spontaneous [Ca2+]i oscillations have demonstrated their implication in the regulation of cell death, axon path finding and synaptic pruning [46 –48]. Therefore, the molecular and functional developmental events reported in this study are likely to be physiologically relevant. Moreover, further investigations on the mechanisms that control [Ca2+]i in neuronal differentiation of stem cells will increase our understanding of activity-dependent mechanisms in the developing CNS or in neural stem cells from the subventricular zone and the dentate gyrus, which persist in human and may allow the development of strategies for preventing neuronal death in degenerative disorders [49].
Specific role of RyR on [Ca2+]i signaling during neuronal differentiation of stem cells
The spontaneous [Ca2+]i elevation in neuronal-differentiated stem cells is mediated by diverse pathways such as intracellular Ca2+ mobilization from Ca2+ stores by RyRs or IP3Rs and through extracellular Ca2+ influx which involves multiple ligand- and voltage-gated channels [50,51]. It has been demonstrated that [Ca2+]i oscillations could be generated by RyRs [52,53]. Our results indicate that neuronal differentiation from CSCs and MSCs could be mediated by RyRs and that RyRs have a key role in promoting spontaneous [Ca2+]i elevation in both cell lines used in this study. Interestingly neurogenesis in ryanodine InhC-treated cultures was significantly reduced, while activation of RyRs with caffeine or ryanodine ExC promoted neurogenesis. The percentage of NEL- and nestin-positive cells is significantly decreased in cultures from ryanodine InhC-treated cultures suggesting that neurogenesis of NPCs from CSCs and MSCs is mediated by Ca2+ signaling through RyRs. This led us to conclude that RyRs affect the generation and proliferation of NPCs and immature neurons. Nevertheless, Ca2+ entry through L-type Ca2+ channels is more efficient than through other types of Ca2+ channels in regulating gene transcription, and thus, in playing a critical role in coupling excitation and transcription in neurons [54]. We also demonstrated that Cav1 channel modulators can affect Ca2+ influx through VGCCs during neuronal differentiation. The number of NEL- or nestin-positive cells increased after exposure to the L-type channel agonist Bay-K or to the depolarizing agent KCl, while the percentage of positive cells decreased after exposure to the L-type channel antagonist nifedipine. Furthermore, in the presence of nifedipine, the NEL- and nestin-positive cells either had a very low [Ca2+]i influx or had no calcium influx in response to membrane depolarization. This let us to hypothesize that cells may express neuronal markers without acquiring a mature neuronal phenotype [55,56]. The marked inhibition of NPC differentiation induced in our model by nifedipine pretreatment suggests that Ca2+ influx through non-Cav1 channels does not play a significant role in neuronal differentiation. The importance of Cav1 channels in neurogenesis is also reflected by their involvement in the regulation of nestin. Thus, the data obtained with antagonists indicate the involvement of L-type channels through ryanodine receptors in the development of functional neuronal cells in CSC and MSC. Further investigations on the detailed pharmacology of these receptors were beyond the scope of the present study. The contribution of each RyR subtype (RyR1, RyR2, and RyR3) could be identified with in vitro differentiation experiments in which expression of these receptors is specifically downregulated by RNA interference (RNAi). This approach could be used to knock down gene expression of specific ryanodine receptors and in different combinations. Further possibilities in studying the functions of RyRs lie in the development of DNA or RNA aptamers as ryanodine receptor subtype-specific inhibitors [57].
Consistent with these results, we found that although caffeine and ryanodine or KCl and Bay-K still evoked low-level [Ca2+]i activity in ryanodine InhC- or nifedipine-treated cultures, this low-level [Ca2+]i activity is not involved in the activity-induced neurogenesis in undifferentiated CSC and MSC. Thus, an antagonist (nifedipine) and an agonist (Bay-K) of L-type Ca2+ channels were effective in inhibiting and promoting neurogenesis, respectively, only in control and ryanodine ExC culture conditions, but not in ryanodine InhC-treated cultures. These results demonstrate that activity-induced Ca2+ influx through L-type Ca2+ channels, if not powered by a CICR mechanism, is not sufficient to promote neurogenesis. Moreover, our data show that RyRs and IP3Rs play, respectively, a major and minor role in mediating the activity-dependent proliferation induced by spontaneous- or neurotransmitter-induced [Ca2+]i elevation, while neurogenesis was mediated by both RyRs and IP3Rs. It is known that neurotransmitters, through activation of their respective receptors, trigger specific patterns of calcium transients inside the cell. This process is a prerequisite for the progress of neuronal differentiation [47]. Various receptor subtypes are activated by the same ligand although coupled to different second messengers. These receptors may induce divergent signaling in some cells while activating convergent signaling in other cells. We have previously shown that for cholinergic [18,19] and purinergic receptors [58] the final pathway that induces neurogenesis or proliferation is that mediated by IP3Rs and RyRs. Thus, pathways dependent on different neurotransmitters or growth factors would mobilize Ca2+ from ER stores through IP3Rs and RyRs. Interestingly, the frequency and amplitude of calcium events can be modulated, directing the acquisition of a defined neuronal phenotype.
Footnotes
Acknowledgments
This work was supported by Instituto do Milenio, CNPq-MCT and Rede Nano, CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico), and FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo), Brazil. R.R.R. and E.L. are CNPq fellows. A.H.K. has postdoctoral fellowship supported by FAPESP. A.A. is acknowledged for suggestions and English editing of the manuscript. Authors would like to thank Guilherme Higa for technical assistance and to L.O. Landeira and L.R. Britto for material and reagents supply.
Author Disclosure Statement
No competing financial interests exist.