
Abstract
Skin-derived precursors (SKPs) from mammalian dermis represent neural crest-related stem cells capable of differentiating into both neural and mesodermal progency. SKPs are of clinical interest because they serve as accessible autologous donor cells for neuronal repair for neuronal intractable diseases. However, little is known about the efficient generation of neurons from SKPs, and phenotypes of neurons generated from SKPs have been restricted. In addition, the neuronal repair using their generated neurons as donor cells has not been achieved. The von Hippel-Lindau protein (pVHL) is one of the proteins that play an important role during neuronal differentiation, and recently neuronal differentiation of neural progenitor cells by intracellular delivery of a synthetic VHL peptide derived from elongin BC-binding site has been demonstrated. In the present study, a synthetic VHL peptide derived from elongin BC-binding site was conjugated to the protein transduction domain (PTD) of HIV-TAT protein (TATVHL peptide) to facilitate entry into cells, and we demonstrate the efficient generation of cells with dopaminergic phenotype from SKPs with the intracellular delivery of TATVHL peptide, and characterized the generated cells. The TATVHL peptide-treated SKPs expressed neuronal marker proteins, particularly dopamine neuron markers, and also up-regulated mRNA levels of proneural basic helix-loop-helix factors. After the TATVHL peptide treatment, transplanted SKPs into Parkinson’s disease (PD) model rats sufficiently differentiated into dopamine neuron-like cells in PD model rats, and partially but significantly corrected behavior of PD model rats. The generated dopamine neuron-like cells are expected to serve as donor cells for neuronal repair for PD.
Introduction
S
Materials and Methods
Isolation of SKPs
Skin-derived precursors were prepared from the back skin of 1–3-day-old Wistar rats. We used a protocol described in detail elsewhere [1,2] with minor modifications. In brief, the dermis of the back skin was dissected and cut into 1–3 mm3 pieces. The tissue fragments were then enzymatically digested with 0.1% trypsin and 0.1% DNase (Sigma, St. Louis, MO). For production of a single cell suspension, they were further mechanically dissociated with a fire-polished Pasteur pipette, and poured through a 40-µm cell strainer (BD Falcon, Bedford, MA). The cells were cultured in growth medium composed of DMEM/F12 (1:1) (GIBCO-Invitrogen, Grand Island, NY) containing 2% B27 supplement (GIBCO-Invitrogen), 20 ng/mL epidermal growth factor (EGF; Upstate Biotechnology, Lake Placid, NY), and 40 ng/mL basic fibroblast growth factor (bFGF; PeproTech EC Ltd., Rocky Hill, NJ) at a density of 1 × 105 cells/mL in a humidified incubator at 37°C with 95% air and 5% CO2. After 7 days, floating clusters (spheres) of cells were centrifuged, mechanically dissociated with a fire-polished Pasteur pipette, and reseeded at the same density in the fresh growth medium for subsequent culture. This procedure resulted in a second generation of spheres. Cells were passaged every 7 days and represented pure SKPs.
Peptide synthesis
By Fmoc (9-fluorenylmethyloxycarbonyl) solid-phase method, we chemically synthesized a peptide corresponding to the 157–171 amino acid sequence of the amino acid region of the pVHL that was demonstrated to be related to neuronal differentiation [21]. The synthesized peptide was liked with the PTD of HIV-TAT protein [TATVHL (157–171)-peptide], thereby facilitating peptide entry into the cells. The FLAG sequence enabled identification of the peptide in cells and tissues. Control peptides containing only the PTD of the TAT protein and FLAG sequence (TATFLAG peptide), or the 104–123 amino acid sequence of the β-domain of the VHL protein [TATFLAGVHL (104–123) peptide] were synthesized [24]. Peptide complexes were also labeled with FITC to observe the cellular internalization of the peptide. These synthesized peptides are listed in Table 1.
T
Neuronal differentiation of SKPs
Sphere-forming SKPs were dissociated into single cells in the medium composed of DMEM/F12 (1:1) containing 2% B27 supplement without growth factors and were plated in 12-well plates coated with laminin and poly-
Immunocytochemistry
Cultured cells were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Cells were immunostained with primary antibodies at 4°C overnight, with secondary antibodies at room temperature for 1 h, counterstained with DAPI, ToPro-3, or YoYo-1 (Molecular Probes, Eugene, OR), and mounted with ProLong Gold (Molecular Probes). Fluorescent images were visualized and captured with a confocal laser scanning microscope (Fluoview FV300; Olympus, Tokyo, Japan). The following primary antibodies were used: rabbit anti-FLAG polyclonal antibody (1:100; Affinity Bioreagents, Golden, CO), mouse anti-Tuj-1 antibody (1:200: R&D Systems, Minneapolis, MN), mouse anti-microtubule-associated protein-2 monoclonal antibody (MAP2; 1:200; Sigma, St. Louis, MO), rabbit anti-neurofilament M polyclonal antibody (NFM; 1:400; Chemicon, Temecula, CA), mouse anti-glial fibrillary acidic protein monoclonal antibody (GFAP; 1:400; Chemicon), goat anti-brain factor 1 polyclonal antibody (BF-1; 1:200, Santa Cruz Biotechnology, San Diego, CA), goat anti-Otx1/2 polyclonal antibody (Santa Cruz Biotechnology), rabbit anti-PAX2 polyclonal antibody (1:200; Zymed Laboratories, South San Francisco, CA), rabbit anti-tyrosine hydroxylase polyclonal antibody (TH; 1:200; Chemicon), rat anti-dopamine transporter monoclonal antibody (DAT; 1:200; Chemicon), rabbit anti-fibronectin (1:400; Sigma), and mouse anti-smooth muscle actin (SMA; 1:200; Sigma). Immunoreactive cells were visualized by using either FITC-conjugated goat anti-mouse IgG (1:200; Sigma), rhodamine-conjugated goat anti-rabbit IgG (1:150; Sigma), or rhodamine-conjugated rabbit anti-rat IgG secondary antibodies (1:150 Sigma). To assess the frequency of different cell types in a given culture, we counted the number of cells immunopositive with a given antibody in 10 to 15 random nonoverlapping visual fields (50–200 cells per field) in each experiment. At least three experiments were performed per condition. The degree of positivity was expressed as the ratio of immunopositive cells to the total number of nuclei stained with ToPro-3, DAP1, or YoYo-1.
Western blot analysis
Cells were lysed with RIPA lysis buffer (0.05 M Tris-HCl, pH 7.4, containing 0.15 M NaCl, 0.25% deoxycholic acid, 1% NP-40, 0.1% SDS, 1 mM EDTA, 1 mM PMSF, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 1 µg/mL leupeptin, and 1 µg/mL aprotinin). Extracted proteins were electrophoresed on 8–15% SDS-PAGE, transferred onto polyvinylidene difluoride membranes, and blots were probed with primary antibodies overnight at 4°C. Immunoreactive bands were visualized by chemiluminescence with ECL-Plus (Amersham Pharmacia). Images were analyzed with LAS-1000 (Fujifilm, Tokyo, Japan), and the density of the bands was determined by using Image Gauge software (Fujifilm). The following primary antibodies were used: anti-Tuj-1, anti-NFM, anti-MAP2, anti-GFAP, anti-TH, anti-DAT, and anti-FLAG.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from SKPs by using TRIzol reagent (Life Technologies Inc., Carlsbad, NY), and then single-stranded cDNA was obtained with oligo-dT primers and avian myeloblastosis virus reverse transcriptase (Takara Bio, Otsu, Japan). PCR was conducted by using the Ex Taq Hot-start version (Takara Bio) with random hexamer primers, all according to the manufacturer’s recommendations. The synthesized cDNA was amplified under the following conditions: initial denaturation at 94°C, 5 min; denaturation at 94°C, 1 min; annealing at 57–59°C, 1.5 min; and extension at 72°C, 1.5 min for 25–35 cycles. The PCR products were then fractionated on 1.2% regular agarose gels and visualized by ethidium bromide staining. To analyze relative expression of different mRNAs, we normalized the amount of cDNA based on the signal from glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. Primer sequences, annealing temperature, and cycle number were as follows: Hes1, sense 5′-CGAGCGTGTTGGGG AAGTA-3′ and antisense 5′-AGTGCGCACCTCGGTGTTA-3′, 57°C, 35 cycles; Hes5, sense 5′-GATGCTCAGTCCCA AGGAGA-3′ and antisense 5′-CGCTGGAAGTGGTAAAG CAG-3′, 57°C, 35 cycles; Neurogenin3 (Ngn3), sense 5′-TGCCTCCAGACGCAATTTAC-3′ and antisense 5′-CTC CACGCGGGACTAGAGTA-3′, 57°C, 38 cycles; NeuroD, sense 5′-CCAGACCTCGTCTCCTTTGTA-3′ and antisense 5′-GGG CTGGTGCAATCAGTTAG-3′, 57°C, 35 cycles; GAPDH, sense 5′-ATTGTCAGCAATGCATCCTGCA-3′ and antisense 5′-AGACAACCTGGTCCTCAGTGTA-3′, 59°C, 25 cycles [25,26].
Analysis of Parkinson’s disease model rats
The procedures for creating the model were described in previous reports [10]. In brief, desipramine (25 mg/kg, intraperitoneally)-pretreated adult male Wistar rats weighing 250–300 g were anesthetized with sodium pentobarbital (40 mg/kg, intraperitoneally) and carprofen (5 mg/kg, subcutaneously) and placed in a stereotaxic apparatus (Narishige, Japan) with the incisor bar set at 3.3 mm below the horizontal. A unilateral lesion was made with 6-hydroxydopamine (6-OHDA) solution (8 µg in 4 µL of 0.1% ascorbate-saline; Sigma) injected in the left medial forebrain bundle (4.4 mm posterior and 1.1 mm lateral to the bregma, and 7.7 mm ventral from the dural surface). All injections were performed with a 10-µL Hamilton syringe at an injection rate of 1.0 µL/min. The rats were challenged with apomorphine (0.1 mg/kg, subcutaneously; Sigma) 2 weeks after the 6-OHDA injection, and the number of rotations was counted over a 30-min period. Only animals showing an average contralateral rotation of greater than 6 turns per minute were included in the experiments. Lesioned rats were divided into three groups: a control group (n = 10) of sham operation; the VHL-SKP group (n = 8), which received SKPs pretreated with 1 µM TATFLAGVHL (157–171) peptide 24 h prior to grafting; and the SKP group (n = 8), which received naïve SKPs. We transplanted 1 × 105 cells stereotactically into the ipsilateral striatum (0.5 mm anterior and 3.0 mm lateral to the bregma, and 4.5 mm ventral from the dural surface). The infusion syringe was placed at the same coordinates unilaterally for sham operation rats, but no fluid was infused. The rotation number was counted for 30 min after apomorphine injection, which carried out at each time point after grafting.
Immunohistochemistry and histology
To evaluate neuronal differentiation of transplanted SKPs in rat brains, immunohistochemical study was performed. Before grafting, SKPs were preincubated with red fluorescence PKH26PCL (Sigma, St. Louis, MO). Transplanted SKPs were divided to three groups: TATFLAGVHL (157–171) peptide-treated cells; nontreated cells; and dead cells, which were quickly frozen and thawed twice. As described, 1 × 105 cells were transplanted into 6-OHDA-lesioned rat striatum. Rats were anesthetized with nembutal (200 mg/kg body weight) and perfused with periodate–lysine–paraformaldehyde solution 8 weeks after the transplantation. Their brains were subsequently dissected and postfixed in the same medium for 2 h, cryopreserved in 30% sucrose for 12 h, and then embedded in Tissue Tek OCT compound (Sakura, Tokyo, Japan). Cryostat coronal sections of 14-µm thickness were prepared for hematoxylin and eosin (H&E) staining and for immunohistochemistry. For immunostaining, sections were incubated with primary antibody overnight at 4°C. The antibodies used were anti-NeuN (1:200; Chemicon), anti-tyrosine hydroxylase (TH; 1:200; Chemicon), and anti-DAT (1:200; Chemicon). Sections were then rinsed and incubated in secondary antibody (FITC-conjugated anti-mouse IgG, 1:200 or rhodamine-conjugated anti-rabbit IgG, 1:150) for 2 h. Counterstaining was done with DAPI. The sections were coverslipped in ProLong Gold (Molecular Probes) and inspected with a confocal laser scanning microscope.
Approval of animal experiment
Wistar rats (Clea, Tokyo, Japan) were used in all experiments and housed in a temperature-controlled room on a 12-h day/night cycle with free access to food and water. All animal experimental procedures were approved by the Institutional Animal Use Committee of Yokohama City University and were in accordance with National Institutes of Health guidelines for care and use of laboratory animals.
Statistics
Numerical data were presented as the mean (%) ± SEM. Factorial analysis of variance applied to each group with pairwise comparison done by the Bonferroni method or Mann–Whitney U-test was noted. To verify whether differences between distinct conditions reached the significance level, we used P < 0.05.
Results
Characterization of multipotent SKPs
Purified populations of floating spheres (ie, SKPs) were obtained by 4 weeks after the initial culture from dermis of neonate rats. The spheres were typically containing >100 cells (Fig. 1A), and the doubling time was 6.9 ± 2.0 days. To assess the characteristics of these isolated SKPs at the passage the floating spheres were dissociated, and the immunocytochemical study was performed for the dissociated cells. At passage 6 of the dissociated cells from the spheres, 99.6 ± 0.5% were fibronectin-positive (Fig. 1B); 70.1 ± 0.3% immunoreactive for nestin, a marker for neural precursors; and 1.4 ± 0.1% immunoreactive for GFAP. We found a very small fraction of SKPs immunopositive for Tuj-1, NFM, or SMA (data not shown). In agreement with a report [1], in the presence of 1–10% FBS and absence of mitogens, the SKPs spontaneously differentiated into neurons (Tuj-1-; MAP2-; and NFM-positive cells), astrocytes (GFAP-positive cells), smooth muscle (SMA-positive cells), and adipocytes after having been cultured for 7 days (Fig. 1D–I). We confirmed by these results that the cultured sphere-formed SKPs were highly enriched in multipotent progenitor cells and that the majority of SKPs underwent a symmetric cell division to produce more SKPs.

Characterization of expanded cells. (
Cellular uptake of the TATFLAG VHL (157–171) peptide
We observed the localization of FLAG-tagged peptide by confocal microscopy. The TAT-mediated distribution was also examined by using anti-FLAG after fixation of the cells in 4% paraformaldehyde. The localization of TATFLAGVHL (157–171) peptide (1 µM) was detected just under the cell surface in vesicles formed at 1–3 h; and diffuse cytoplasmic or nuclear localization was seen at 6 h, followed by complete nuclear translocation by 24 h. Our measurements showed that TAT complexes were transferred into virtually all of the cells in rapid fashion and that the complexes were subsequently targeted to the nucleus. The TAT-mediated peptide internalization was dose-dependent (Fig. 2).
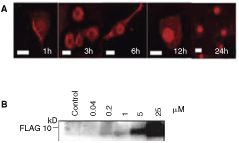
Intracellular delivery of protein transduction domain (PTD)-linked VHL (157–171) peptide into cultured skin-derived precursors (SKPs). (
TATFLAGVHL (157–171) peptide induces the dopaminergic differentiation of SKPs
After treatments with TATFLAGVHL (157–171) peptide, TATFLAGVHL (104–123) peptide, or TATFLAG peptide (1 µM), and subsequent culture for 7 days in neuronal differentiation medium, SKPs were then evaluated for their neuronal differentiation by morphological and immunophenotype analyses. In the morphological analysis, neurite outgrowth was assessed. Twenty-four hours after the treatment with TATFLAGVHL (157–171) peptide, SKPs were started to be induced to have neurite outgrowth, and 72 h after the treatment 44.1 ± 5.0% of cells showed neurite outgrowth and the formation of network was observed. In the immuocytochemical study using NFM, the percentage of NFM-positive cells was significantly higher in the TATFLAGVHL (157–171) peptide-treated cells (40.4% ± 4.4%) than in the control peptides-treated cells (TATFLAGVHL (104–123), 12.0 ± 3.2%; TATFLAG, 8.9 ± 1.5%, P < 0.01). This result indicated that TATFLAGVHL (157–171) peptide induced SKPs to differentiate into cells that expressed NFM. Furthermore, expressions of basic helix-loop-helix (bHLH) transcription factors, known to be involved in the regulation of proliferation and lineage determination of NSCs, were examined in TATFLAGVHL (157–171) peptide-treated SKPs and control peptide-treated cells. Hes1 and Hes5 are essential for maintenance in stem cell fate and proliferation [25]. RT-PCR revealed that mRNA level of Hes1 was highly expressed in TATFLAGVHL (104–123) peptide-treated cells and TATFLAG peptide-treated cells, but reduced in those in TATFLAGVHL (157–171) peptide-treated cells. Moreover, we found high expression of mRNAs of proneural bHLH (Ngn3 and NeuroD) in TATFLAGVHL (157–171) peptide-treated cells. Hes5 was not detected under any condition. These results demonstrate that the 157–171 amino acid sequence of pVHL mediated the switch of the precursor cell fate into the neuronal lineage (Fig. 3).
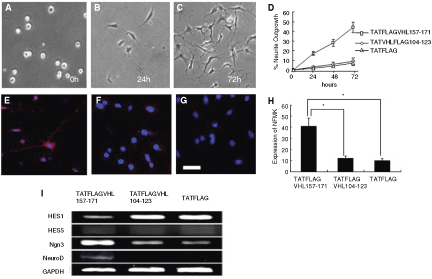
Morphological and immunocytochemical studies for peptide-treated skin-derived precursors (SKPs). (
In addition, the phenotype of TATFLAGVHL (157–171)- treated cells was examined with immunocytochemical study and Western blot analysis. The immunocytochemical study for TATFLAGVHL (157–171) peptide-treated cells showed the high rates of expressions of midbrain markers: Otx1/2 (15.2 ± 1.7%), PAX2 (27.2 ± 2.5%), DAT (41.0 ± 4.3%), and TH (32.8 ± 3.1%) but low rate of expression of BF1 (5.9 ± 0.7%), caudal telencephalon maker. On the other hand, the studies for TATFLAGVHL (104–123) peptide-treated and TATFLAG peptide-treated cells showed the low rates of expressions of midbrain markers: Otx1/2 (6.2 ± 0.6%; 5.7 ± 0.5%); PAX2 (5.6 ± 0.6%; 4.7 ± 0.5%); DAT (6.1 ± 0.7%; 5.5 ± 0.6%); and TH (5.8 ± 0.7%; 4.0 ± 0.5%), respectively. In the study for the cells cultured in dopaminergic neuron differentiation media containing FGF-8 and Shh, the rates of expressions of midbrain markers: Otx1/2 (8.1 ± 1.3%); PAX2 (13.9 ± 1.7%); DAT (19.3 ± 2.6%); and TH (14.2 ± 1.8%) showed significantly higher than those of TATFLAGVHL (104–123) peptide-treated and TATFLAG peptide-treated cells, but significantly lower than those of TATFLAGVHL (157–171) peptide-treated cells.
Western blot analysis revealed that the protein levels of NFM, MAP2, Tuj-1, TH, and DAT were markedly increased in the TATFLAGVHL (157–171) peptide-treated cells. In contrast, the level of GFAP was markedly reduced. These results indicated that the TATFLAGVHL (157–171) peptide-treated cells differentiated to the cells showing dopaminergic phenotype (Fig. 4).
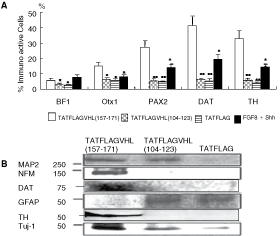
Phenotype study for peptide-treated and control skin-derived precursors (SKPs). (
Behavioral and histological evaluation after transplantation of SKPs into PD model rats
To explore the possibility of the SKPs could function and survive in vivo, we grafted cells into the striatum of unilateral 6-OHDA-lesioned rats, commonly used model of PD. Two weeks after the 6-OHDA lesion, the rats were depleted of dopaminergic innervations in the ipsilateral striatum and exhibited a characteristic rotation behavior as a response to the apomorphine challenge. Lesioned rats were divided into three groups: (1) control group of sham operation (n = 10), (2) VHL-SKP group (n = 8) engrafted with TATFLAGVHL (157–171) peptide-treated SKPs, and (3) SKP group (n = 8) engrafted with naïve SKPs. A functional analysis of apomorphine-induced rotation behavior was revealed that rats in SKP group showed no remarkable recovery compared with sham-operated rats at any time point. In contrast, rats in VHL-SKP group showed a slight but significant improvement over 4 week after the transplantation (P < 0.05). The rotations decreased in the VHL-SKP group from an average of 9.35 ± 0.75 times/minute at baseline to 7.55 ± 0.84 (20% decrease), 6.85 ± 0.65 (27%), 6.46 ± 0.67 (31%) at 4, 8, and 12 week, respectively. Meanwhile, the rotations increased from 9.35 ± 0.52 at baseline to 10.0 ± 1.41 at 12 week in the control group and from 9.05 ± 0.94 to 11.0 ± 1.26 in the SKP group. To evaluate the survival and the morphological maturation of grafted SKPs in the striatum of 6-OHDA-lesioned rats, we prelabeled the cells with red fluorescence PKH26PCL (Sigma, St. Louis, MO) before grafting. The animals were killed for histological analysis 8 weeks after engraftments. No tumor formation by the grafts was found following hematoxylin and eosin staining (data not shown). Immunohistochemical analysis of sections by confocal microscopy revealed that ∼5% of the PKH-labeled transplanted cells survived in the PD rats. In the VHL-SKP group, PKH-labeled cells showed high positive rates of NeuN (27.5 ± 3.5%) as well as TH (38.7 ± 4.6%) and the sprouting of dopaminergic neurons surrounding the PKH-labeled cells was enhanced, while in the sections of naïve SKP group PKH-labeled cells showed low positive rates of NeuN (7.2 ± 0.8%) as well as TH (4.7 ± 0.5%) and in section of PKH-prelabeled frozen and thawed dead cells or fragments did not show positive for NeuN (Fig. 5).
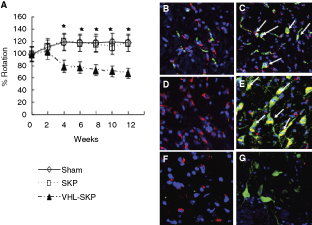
Integration of TATFLAGVHL (157–171) peptide-treated skin-derived precursors (SKPs) engrafted into the striatum of Parkinson’s disease (PD) rats and the behavior of the treated SKPs-engrafted PD rats. (
Discussion
SKPs are the ideal precursor cell populations that could be derived in an autologous fashion from small amounts of accessible tissue biopsies and are pluripotent somatic stem cells capable of differentiating into both neural and mesodermal progency. Although SKPs have potential to differentiate into neural cells, the phenotype of the neural cells differentiated from SKPs has been restricted to peripheral neuron or oligdendrocyte-phenotype in the differentiating conditions that were previously reported. Before cell transplantation for regeneration-aimed cell-based therapy for neuronal disease, neuronal differentiation of stem cells is fundamental, since untreated naïve stem cells scarcely differentiate to neurons in engrafted neural tissues. However, differentiation to another neuron phenotype in SKPs and an efficient generation of neurons from SKPs have not been demonstrated. Therefore, a novel neuronal differentiation method has been required for differentiation to another neuron phenotype in SKPs and an efficient generation of neurons from SKPs. Recently, we demonstrated neuronal differentiation of NPCs with intracellular delivery of an oligopeptide derived from pVHL [21]. Then, in the present study we proposed a novel method using intracellular delivery of PTD-linked oligopeptide derived from VHL for neuronal differentiation in SKPs. For intracellular protein transduction, intracellular delivery of cell-penetrating protein conjugated with PTD [22,23] or protein–lipid complex [26] is superior to gene transfer, a most user-friendly method, which is accompanied with problems such as troublesome handling, toxicity, and infectious risk in viral vectors. In addition, we delivered not a full-length protein but an oligopeptide of functional domain responsible for the function of the full-length protein, because an oligopeptide delivered intracellularly can have effect similar to those obtained by transfer of the gene encoding the full-length protein [21,27,28], and an oligopeptide is chemically synthesized with ease and its delivery is superior to that of the full-length protein because of smaller molecule.
In the present in vivo study, SKPs with specific induction using TATFLAGVHL (157–171) peptide were shown to be differentiated into dopamine neuron-like cells in vitro and after transplanted into PD model rats. They also relieved their apomorphine-induced asymmetric motor behavior by 31% in this study. On the other hand, our transplantation experiment has also shown that untreated naïve SKPs were unsatisfactorily differentiated into dopamine neuron-like cells and insufficient to recover the behavior of the PD model rats, which may reflect to be in the absence of an inductive signal and/or the presence of an inhibitory signal. These results suggested that it was thus fundamental to SKPs to be inducted into neuronal differentiation prior to grafting for cell transplantation therapy for PD, and our presented approach would contribute to therapy for PD.
In conclusion, dopamine neuron-like cells are efficiently generated from SKPs with intracellular delivery of the TATFLAGVHL (157–171) peptide, and also the engrafted TATFLAGVHL (157–171) peptide-delivered SKPs restore the physiological function in 6-OHDA-lesioned PD model rats. This study provides a promise of clinical application of a new autologous donor cell, derived from the readily accessible skin, for the cell transplantation therapy for patients with PD.
Footnotes
Acknowledgments
This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Basic Research No. 16390415) and Yokohama General Medical Foundation. We would like to thank Dr. Hidemitsu Sato for discussion and Dr. Masaya Baba for providing adenovirus vector, as well as Ms. Kyoko Arai and Ms. Teruyo Watanabe for their technical assistance.