
Abstract
Although the pluripotent and proliferative capacity of embryonic germ (EG) cells is thought to be equivalent to that of embryonic stem (ES) cells, there has been far less attention focused on the potential use of EG cells for applications in developing novel strategies of tissue transplantation in the treatment of degenerative diseases. In this study, EG cells were derived from primordial germ cells (PGCs) of genital ridges of 4-day-old chicken embryos. These cells satisfied the criteria previously used for defining chicken EG cells by using the expression of markers characteristic to ES cells. When injected subcutaneously, chicken EG cells could form teratomas that enable differentiation into a wide range of tissue types of all three primary cell lineages including neural cells, cartilage, forming bone, adipocytes, blood vessels, smooth muscle, and secretory epithelia in the recipients. Furthermore, cells in embryoid bodies (EBs) expressed lineage-specific markers of three germ layers and could be induced to differentiate into more advanced stages of various committed cell types, including dopamine and cholinergic neurons, astrocytes, oligodendrocytes, adipocytes, and hepatocytes, which were demonstrated by immunocytochemical staining or RT-PCR analysis. These findings support the multilineage differentiation capability of chicken pluripotent EG cells, thus confirming the presumption that chicken embryos may be used as a potential model for better understanding the mechanisms of tissue-specific differentiation and regeneration that will help to devise strategies based on the transplantation of stem cell-derived tissues for restoring function to damaged or diseased tissues.
Introduction
A
The pluripotency of ES cells is evident from three main features: (1) these undifferentiated cells can be injected into the blastocyst to form all cell types in the chimeric progeny; (2) subcutaneous injection of these cells into syngenetic animal induces teratomas that may include cells of endodermal, ectodermal, or mesodermal origin; (3) In vitro differentiation via embryoid body (EB) formation, which results in collections of precursors and differentiated cells from a wide variety of lineages. During the last decade, EG cell lines that share most of these characteristics have been mostly reported for the mouse [9,10] and human [5,11]. Chicken embryo has become one of the most versatile experimental systems available [12] and served as a model for regenerative medicine [13] due to several new technologies including the isolation of embryonic stem cells, novel methods for transgenesis, and the sequencing of the chick genome. Since the report of chicken EG cells derived from PGCs harvested from the undifferentiated gonad by Park and Han [14], increasing interest in these cells has been stimulated by the potential use of EG cells in production of chicken chimaeras [14 –17]. However, equivalent reports on in vivo teratoma formation and in vitro differentiation into desired cell types via EBs formation have, in comparison, been lacking.
In our previous study, the quantity of chicken PGCs was amplified by epidermal growth factor via activation of Ca2+/PKC involving NFKB1 signaling pathway in culture [18]. In this study, we derived EG-like cells from chicken PGCs and investigated the potentials of differentiation. These cells exhibit features similar to those of ES cells such as typical growth morphology of tightly compact multicellular colonies, strong positive staining for periodic acid-Schiff (PAS), stage-specific embryonic antigens (SSEA-1, SSEA-3, and SSEA-4) antibodies, the expression of the pluripotency-associated genes such as cPouV, cNanog, and Sox2. At the same time, these EG-like cells were essentially like ES cells that could form teratoma in vivo and be capable of differentiation into various desired multilineage cell types in vitro containing mature neural cells, adipocytes, and hepatocytes, thus is certainly an experimental model to be considered when aiming to develop a regenerative therapy.
Materials and Methods
Isolation and maintenance of EG-like cells in culture
Fertilized Arbor Acres broiler chicken eggs were incubated at 38.5% and 60% humidity for 4 days in an egg incubator. PGCs were prepared according to a previous study [19]. For primary culture, cell suspension containing both PGCs and somatic cells was seeded onto gelatin-treated 35-mm culture plates (Costar, Corning Inc., Lowell, MA) at a density of 1 × 106/well in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 5% fetal calf serum (FCS), 10 ng/mL leukemia inhibitory factor (LIF, StemCell Inc., Vancouver, British Columbia, Canada), 10 ng/mL human basic fibroblast growth factor (bFGF, Peprotech, Rocky Hill, NJ), 0.1 mM minimum essential medium (MEM) nonessential amino acids (Sigma, St. Louis, MO), 0.1 mM 2-mercaptoethanol (Invitrogen, USA), 2 mM
Characterization of EG-like cells
After three passages, the presumed chicken EG-like colonies were taken for characterization. PAS staining was conducted according to the method described by Tang et al. [19]. In brief, cultured EG-like colonies were fixed in 4% formaldehyde for 10 min. After rinsing in PBS, the cells were then immersed in periodic acid solution for 5 min and subsequently treated with Schiff’s reagent for 15 min. To detect expression of stem cell markers, EG-like colonies were examined for expression of the stage-specific SSEA-1, SSEA-3, and SSEA-4 by immunocytochemical staining, as well as the transcription factors cPouV, cNanog, and Sox2 by RT-PCR.
To induce the formation of teratoma, 7-day-old chicks were obtained from a commercial hatchery and fed with cyclosporine (5 mg/kg BW, Sandoz, Austria) known as immunosuppressive agent to dampen down the body’s immune reaction. EG colonies were collected and treated with 0.25% trypsin–EDTA to achieve single-cell suspension. About 1 × 106 single cells in 200 µL PBS were injected subcutaneously into axilla region of chicks. After 5 weeks, chicks were sacrificed and emerging tissue materials were dissected. Tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Sections (5 µm) were placed on slides (ProbeOnPlus, Fisher Scientific, Pittsburgh, PA) and stained with hematoxylin and eosin.
Induction of EG cell differentiation in vitro
Prior to induction of EG cell differentiation, colonies in culture were picked up and dissociated into single cells, transferred onto gelatin-coated plates for a 30-min period to remove contaminating somatic cells, and then transferred onto Ultra Low Attachment plates (StemCell Inc., Canada) to allow their aggregation and prevent adherence to the plate. Usually about 106 EG cells were incubated in each 35-mm plate accompanied by the withdrawal of LIF and 2-mercaptoethanol from the medium. During this period, the cells aggregated to form EBs, which were then plated onto gelatin-coated culture dishes and treated with growth factors that can be used to induce or allow the expansion and survival of desired cell types.
For the generation of the neural cells, EBs were plated onto gelatin-coated tissue culture plates at Day 4. Neural progenitors were induced in serum-free medium containing 10−6 M retinoic acid (RA), 2 mM glutamine, 5 mM HEPES, 25 µg/mL insulin, 100 µg/mL transferrin, and 30 nM sodium selenite for 2 days. To induce mature neural cells, these progenitors were continually cultured for 7 days. Adipogenic differentiation was induced by culturing 7-day-old EBs in medium supplemented with 10% FBS, 10 µg/mL insulin, 1 µM dexamethasone, 0.2 mM indomethacin for 10 days. The differentiated cells were washed with PBS and fixed with 4% paraformaldehyde for 30 min at room temperature. Then the cells were incubated with 0.3% Oil Red O (Sigma-Aldrich, St. Louis, MO) in 60% isopropanol for 30 min. Fetal chicken liver-derived cells were prepared as adherent cells from 7-day-old chicken embryos, after which 14-day-old EBs were plated onto the gelatin-coated culture dishes and incubated in the liver cells-conditioned medium. Differentiated cells growing out from the EBs were analyzed by immunocytochemical staining and RT-PCR analysis for expression of the specific markers.
Immunocytochemical staining
The expression of each antigen was examined in separate experiments at least three times. Cells were fixed with 4% paraformaldehyde for 15 min at room temperature and washed three times with PBS. Next they were treated in 3% H2O2 for 15 min and washed three times in PBS. Nonspecific binding was blocked by a 30-min treatment in 15% normal goat serum. The cells were incubated with primary antibodies at 4°C overnight. Sources and dilution of primary antibodies were as follows: mouse anti-SSEA-1, anti-SSEA-3, and anti-SSEA-4 (1:50; Chemicon, Temecula, CA), rabbit anti-neuron-specific enolase (NSE), anti-glial fibrillary acidic protein (GFAP), anti-2′,3′-cyclic nucleotide 3′ phosphodiesterase (CNP), anti-tyrosine hydroxylase (TH), anti-choline acetyltransferase (ChAT, 1:100, Boster, Wuhan, China), anti-albumin (ALB, 1:100, Bioss, Beijing, China). After three-time washes in PBS, cells were incubated with horseradish peroxidase-conjugated IgG (1:200) or FITC-conjugated goat anti-rabbit IgG (1:64, Boster, Wuhan, China) for 1 h at 37°C. After washing, the immunoreaction was detected by using DAB system or a fluorescence microscope. For negative controls, only the primary antibody was replaced with normal serum.
RT-PCR analysis
Total RNA was extracted from primary PGC colonies, undifferentiated EG cells, EBs, or differentiated cells with the UNIQ-10 Column RNA isolation kit. The cDNA was synthesized from total RNA using a AMV First-Strand cDNA synthesis kit with oligo(dT)18 primers according to the manufacturer’s instructions (Sangon, Shanghai, China). PCR was subsequently carried out on 1 µL cDNA template for 30–40 cycles with primers as listed in Table 1. As an internal control, β-actin expression was assayed using the same reactions. The PCR products (5 µL) were analyzed by electrophoresis on 1.7% agarose gel containing 0.1 µg/mL ethidium bromide and photographed on a Tanon Gel Imaging system (Tanon, Shanghai, China). The electrophoresis band intensities of the PCR products were quantified using Tanon Gel Imaging system. Mean mRNA expression levels normalized against β-actin levels detected in the same cDNA sample were presented in absolute integrated optical density. Each value was analyzed for statistical difference by SAS software.
P
Results
Culture and characterization of EG cells
In the present study, the PGCs were plated together with their surrounding somatic cells as primary culture. The somatic cells attached to the surface of the culture dish and used directly as feeder cells. PGCs attached to the surface of the feeder layer firmly pack together into large multicellular colonies 5 days after seeding (Fig. 1A). Numerous putative EG colonies with ES cell-like morphology could be observed after subculture. Throughout the culture period most cells within the colonies continued to be PAS positive (Fig. 1B and C) and stained strongly with antibodies against SSEA-1, SSEA-3, and SSEA-4 (Fig. 1D–G). Besides the immunocytochemical identification, these EG cells were further characterized by teratoma formation in vivo (Fig. 1H–O) and induced multilineage differentiation via EB formation in vitro (Fig. 2).
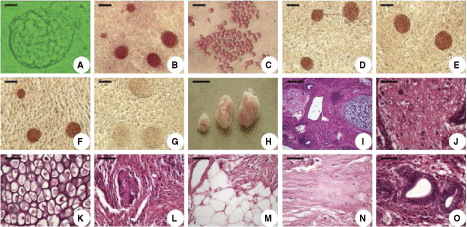
Characterization of colony-forming chicken embryonic germ (EG) cells and morphology of EG cell-derived teratomas. (

Characterization of the differentiated cells from chicken embryonic germ (EG) cells in vitro. (
There was intensive expression of the PGC marker genes, such as chicken vasa homolog (Cvh), deleted in azoospermia-like (Dazl), and chicken dead end homolog (CDH) (Fig. 3A). Therefore, it was determined that most of the cells in primary formed colonies after 5 days in culture were PGCs. Additionally, expression of cPouV, cNanog, and Sox2, which are proved to be critical for stem cell self-renewal and pluripotency, were also detected in EG-like colonies (Fig. 3B).
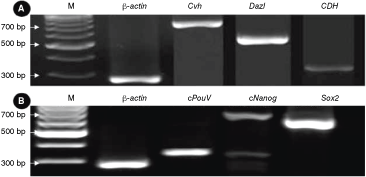
Analysis of mRNA expression by RT-PCR. (
Formation of teratoma in vivo
Chicken EG cells were capable of forming teratomas after subcutaneous injection into the immunosuppressed chicks (Fig. 1H). After 5 weeks, the teratomas were recovered and processed for histological analysis. The amount of differentiated tissues varied among individual teratomas. Each teratoma contained a broad variety of tissues (Fig. 1I), including neural cells, cartilage, forming bone, adipocytes, blood vessels, smooth muscle, and secretory epithelia (Fig. 1J–O). There was no teratoma formed in chicks without immunosuppression.
Formation of EBs in vitro
When EG cells were taken into suspension culture, the cells grew into compact aggregates and became simple EBs within 2–5 days (Fig. 2A and C), then turned to cystic EBs with the formation of a central cavity (Fig. 2B and D). EG cell-derived EBs were replated onto gelatin-coated plates under different conditions in an attempt to promote multilineage differentiation. In addition, in order to confirm that this described differentiation surely came from EG cells instead of a contaminating population of progenitor cells in PGC colonies from which the EG cells originated, our study showed that under standard culture conditions, the expression of progenitor markers including nestin, peroxisome proliferation-activated receptor gamma (PPARγ), and α-fetoprotein (AFP) was not detected in EG cells by RT-PCR analysis (Fig. 4).
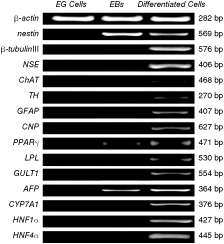
RT-PCR analysis of cell-specific gene expression during chicken embryoid body differentiation in vitro. Abbreviations: EB, embryoid body; EG, embryonic germ.
Induced differentiation of EG cells into neural cells
To elucidate the capacity of EG cells to generate neural cells, we examined by RT-PCR the expression of the neural progenitor marker and the mature neural cell makers during the differentiation process (Fig. 4). Results showed that RA treatment together with serum-free culture condition significantly increased levels of neural-specific gene expression (Fig. 5A). After additional 7 days in culture, differentiated cells grew out from EBs into monolayer. Cells possessing typical neuronal morphology were fixed and proved to be NSE, TH, ChAT, GFAP, and CNP positive by immunocytochemical staining (Fig. 2E–I). RT-PCR analysis also showed that after induction nestin was down-regulated, whereas NSE, β-tubulinIII, ChAT, TH, GFAP, and CNP became positive, which displayed mature neuron, astrocyte, and oligodendrocyte differentiation (Fig. 5A).
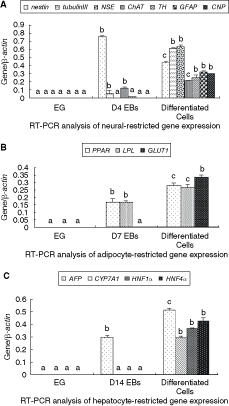
The integrated optical density (IOD) ratio of cell-specific genes versus β-actin electrophoresis bands at different differentiation stages. (
Induced differentiation of EG cells into adipocytes
The preadipocytes marker PPARγ was already detected in 7-day-old EBs, while glucose transporter protein-1 (GLUT1) was expressed at low levels (Fig. 4). For the generation of mature adipocytes, 7-day-old EBs were incubated in adipocytes induction medium for 10 days. The accumulation of lipid vacuoles in the cells was first detected 5 days after the addition of adipogenic induction medium and determined by staining for Oil Red O 10 days after induction (Fig. 2J). As differentiation progressed, expression of PPARγ, GLUT1 was significantly increased (Fig. 5B). Mature adipocytes marker gene lipoprotein lipase (LPL) was detected 10 days after induction, thereby confirming the mature adiogenic differentiation (Fig. 5B).
Induced differentiation of EG cells into hepatocytes
The AFP mRNA expression was detected in 14-day-old EBs. However, the expression of cytochrome P450 7a1 (CYP7A1), hepatocyte nuclear factor 1α (HNF1α), and hepatocyte nuclear factor 4α (HNF4 α), possible markers for embryonic endoderm-derived mature hepatocytes, were only observed after a co-culture with fetal liver cell-derived conditioned medium (Figs. 4 and 5C). Immunocytochemical staining together with histochemical results demonstrated a high ratio of ALB and PAS positive cells 10 days after induction of hepatocyte differentiation, thus showing that co-culture of EG cells with liver cells-derived conditioned medium guided the differentiation toward mature and functional hepatocytes (Fig. 2K and L).
Discussion
Considerable effort is currently focused on understanding and controlling stem cell differentiation for the purpose of deriving cells for tissue replacement therapy. Studies using chicken PGCs or its derived EG cells have been used extensively to novel system for the production of transgenic chickens. Since EG cells are highly similar to ES cells in their characteristics, they may provide as a experimental model of pluripotent stem cells for tissue replacement therapy. Here we isolated PGCs from the genital ridges of 4-day-old chicken embryos and found that they have high efficiency in further proliferation and colony formation. The isolated PGCs grew in large, multilayered colonies, and these colonies were densely packed with an obvious border after primary culture. However, the morphology of the primary formed PGC colonies was quite different from that of the embryonic blood or the gonads derived PGCs [14,16]. This might be resulted from the differences of physiological and differentiation aspects of the PGCs among different developmental stages. During the last decade, Cvh, Dazl, and CDH have been found expressed exclusively in chicken PGCs [16,20,21]. Therefore, to trace the origin of the colonies, the primary formed colonies were subjected to RT-PCR analysis after 5 days in culture for expression of these PGC-specific markers.
Upon previous methods, PGCs can promote the colonic formation and the conversion to EG cells with the strict cell culture conditions [14,15]. In this study, when the primary PGC colonies were separated and reseeded in the culture, new tightly compacted multicellular colonies with distinct boundaries that were similar to that of mouse ES and EG colonies were produced within 3–5 days of subculture. The newly isolated EG-like cells could be maintained for >2 months in vitro in this study. Besides the morphological evaluation of EG-like colonies, they also satisfied the criteria previously used to define chicken EG cells including positive staining for PAS, SSEA-l, SSEA-3, and SSEA-4. cPouV, cNanog, and Sox2 are known as three critical transcription factors that act cooperatively to maintain pluripotency in both mouse and human embryonic stem cells [22]. Studies on expression of these pluripotent markers in chicken ES cells have made some progress recently [23]. However, the expression of these critical transcription factors in chicken EG cells was first detected in the present experiment. Since the potency of the EG cells to generate chimeras and donor-derived offspring was already demonstrated by several studies [15 –17], we did not repeat this experiment. Alternatively, we demonstrated the ability of the EG cells to form teratomas in recipient when injected subcutaneously, which also manifested their potency of multilineage differentiation in vivo.
Over the past few years, a great deal of effort has been directed at defining conditions, which promote the differentiation of ES cells into functional neurons in the hope that they can be used in cell replacement therapies to treat neurodegenerative diseases. More reports issued from EG cells by using EB strategy or high-density culture broaden the methods of neuronal induction. All-trans-retinoic acid is well known as the biologically active form of vitamin A and has been shown to play an important role during embryogenesis [24]. More reports showed that exposure to high concentration of RA strongly drove neural induction [25,26]. Previous studies have displayed neural differentiation tendency of chicken EG cells [14]. Our experiments further investigated the induction of neural differentiation of chicken EG cells by treatment with RA to mature neurons and glial cells in vitro. To demonstrate the maturation of neurons in our culture system, we analyzed the expression of NSE and β-tubulinIII, markers of mature neurons, as well as the neurotransmitters including TH, an enzyme required for the biosynthesis of dopamine, and ChAT, the rate-limiting enzyme of acetylcholine (Ach) synthesized in cholinergic neurons. In addition, glial differentiation was also confirmed by immunostaining and mRNA expression for the astrocyte marker GFAP and the oligodendrocyte marker CNP.
Adipogenesis is a complex process that is tightly controlled by positive and negative stimuli, including a variety of hormones and nutrients [27,28]. In vitro, a combination of several soluble factors such as insulin, indomethacin, and dexamethasone is commonly used to induce differentiation of preadipocytes though the exact mechanisms of action are not fully known [29,30]. Adipogenic differentiation was accompanied by the markedly increased expression of PPARγ, which is a key transcription factor in the early stages of preadipocyte differentiation [31]. The differentiation of cells to mature adipocytes in the differentiation medium was verified by observed increases in LPL and GLUT1 expression, which were adipose tissue-specific genes and played an important role in the adipocyte differentiation and mature process [32].
A number of studies have examined the differentiation of mouse and human ES cells to hepatocyte-like cells and have obtained various extents of hepatic induction, with the expression of specific markers and functional enzymes of hepatocytes [33 –35]. As the process of gestation proceeds from midgestation to birth, the fetal liver increases dramatically in size and switches to a metabolic organ from a hematopoietic organ at around the time of birth. Fetal liver-derived cells were considered capable to provide a conductive environment for ES cells to differentiate along a hepatic lineage [33,36]. As an alternative resource of pluripotent stem cells, it is still largely obscure that whether chicken EG cells can differentiate into mature hepatocytes. In the present study, we co-cultured undifferentiated EG cells with liver-conditioned medium and found early expression of AFP, a hepatocyte-related marker, which remained throughout the experimental period. Cells at end of the differentiation resembled hepatocytes morphologically and expressed a repertoire of genes expressed in mature hepatocytes such as CYP7A1, a definitive marker for endoderm-derived mature hepatocytes, HNF1 α, and HNF4 α, two liver-enriched transcription factors. Further studies on PAS and ALB staining suggested that diffusible factors from fetal liver cell-conditioned medium seemed to be sufficient to stimulate the induction of hepatic differentiation into glycogen and albumin-producing hepatocytes from chicken EG cells.
Although the chicken embryo has long been used as a model for developmental biology, its potential use as an experimental model for the repair and regeneration of adult tissues is often overlooked. Through this study we successfully isolated positive cells for chicken PGC-specific markers from genital ridges. The positive cells proliferated to form colonies on somatic cells during primary culture. Firmly packed colonies with ES cell-like morphology could be observed after subculture. These cells showed important features of pluripotent stem cells, especially the expression of the pluripotency-associated genes cPouV, cNanog, and Sox2. The capacity of teratoma formation and their multilineage differentiation potency into mature neuron, glial cells, adipocytes, and hepatocytes fully provide evidence that chicken PGC-derived EG cells possess the pluripotency to differentiate into precursor cell lineages of all three germ layers and then into mature cells after the directed induction methods in vitro, thus making them a potential experimental model for further studies of the cellular mechanisms of tissue-specific differentiation and regeneration, which will help to devise strategies for restoring function to damaged or diseased tissues in animals or humans.
Footnotes
Acknowledgments
This study was supported by the Chinese Ministry of Education (NCET-05-0514), the National Natural Science Foundation of China (30871843), and Zhejiang Provincial Natural Science Foundation (R305035). We thank Drs. Xun Tan and Jiusheng Wu, and Minli Yu (Zhejiang University) for help in this study.