
Abstract
Recent evidence suggests that sphingosine 1-phosphate (S1P) regulates self-renewal of human embryonic stem (ES) cells and differentiation of mouse embryoid bodies (derived from mouse ES cells) to cardiomyocytes. We have investigated the role of S1P in regulating ERK-1/2 signaling in mouse ES cells. In this regard, we found that both mouse ES-D3 and CGR8 cells express S1P1, S1P2, S1P3, and S1P5 but lack S1P4. The treatment of ES cells with S1P induced the activation of ERK-1/2 via a mechanism that was not mediated by S1P1, S1P2, or S1P3. This was based on: (i) the failure of S1P1, S1P2, or S1P3 antagonists to inhibit S1P-stimulated ERK-1/2 activation and (ii) the failure of SEW 2871 (S1P1 receptor agonist) to stimulate ERK-1/2 activation. The treatment of ES cells with phytosphingosine 1-phosphate (phyto-S1P), which we show here is an agonist of the S1P5 receptor, stimulated ERK-1/2 activation. These findings therefore suggest that S1P5 may mediate the effects of S1P in terms of regulating ERK-1/2 signaling in ES cells. The S1P-dependent activation of ERK-1/2 was sensitive to inhibition by pertussis toxin (uncouples the G-protein, Gi from GPCR), bisindolylmaleimide I (PKC inhibitor), and PP2 (c-Src inhibitor), but was not reduced by LY29004 (PI3K inhibitor) suggesting that S1P uses Gi-, PKC-, and c-Src-dependent mechanisms to activate the ERK-1/2 pathway in ES cells.
Introduction
E
Sphingosine 1-phosphate (S1P) binds to a family of five G-protein-coupled receptors (GPCR) termed S1Pn (where n = 1–5) [12] that are differentially coupled to heterotrimeric G-proteins (Gi, Gq, and G12/13) to regulate various effectors, such as MAP kinases, linked to diverse cellular processes such as proliferation, cell survival, and differentiation. S1P and platelet-derived growth factor (PDGF), a potent mitogen, have been found to promote the differentiation of mouse ES cells to cardiomyocytes [13]. Using a defined serum replacement protocol to identify specific growth factors promoting cardiac development, stimulation of EB with either PDGF-BB or S1P resulted in a 2.6-fold enhancement of cardiomyogenesis, quantified by the expression of cardiac-specific myosin heavy chain alpha and beta (αβMHC) and beating activity. S1P and PDGF have also been demonstrated to promote pluripotency in human ES cells [14]. Culture of human ES cells in the presence of S1P or PDGF reduced spontaneous differentiation compared with controls. Furthermore, co-incubation with S1P and PDGF resulted in greater inhibition of spontaneous differentiation. This effect was reduced when cells were treated with pertussis toxin or a MAP kinase inhibitor suggesting that interruption of differentiation is mediated via a GPCR and involves activation of extracellular signal-regulated kinases (ERK). More recently, Wong et al. [15] reported that S1P and PDGF have an antiapoptotic effect in human ES cells and that this protection is contingent on the activation of ERK-1/2. We have therefore investigated S1P signaling to ERK-1/2 in mouse ES cells and its modulation by PDGF.
Materials and Methods
Regents
All biochemicals were obtained from Sigma Chemical Company (Poole, UK) unless otherwise stated. Cell culture supplies and Lipofectamine™ 2000 were obtained from Invitrogen (Paisley, UK). S1P was purchased from Avanti Polar Lipids (Alabaster, USA), G418 from Merck Biosciences Ltd (Nottingham, UK), and Leukaemia Inhibitory Factor (LIF) from Millipore (Livingston, UK). Antibodies used were from suppliers as follows: anti-S1P5 (# sc-25493), anti-phosphorylated ERK-1/2, and anti-HA from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA) and anti-ERK-2 from BD Biosciences (Oxford, UK). [Methyl- 3H]Thymidine (25 Ci/mmol) was purchased from G.E. Healthcare (Little Chalfont, UK). SB649146 was a kind gift from Glaxo Smith Kline (USA), while JTE013 and CAY10444 were purchased from Tocris Bioscience (Bristol, UK) and Cayman Europe (Tallinn, Estonia), respectively. Phytosphingosine 1-phosphate (phyto-S1P), DM95, and DM97 were obtained by selective phosphorylation of the corresponding N-Boc precursors (Mormeneo et al., unpublished).
Cell culture
ES-D3 cells were cultured on 0.1% (w/v) gelatin/phosphate-buffered saline (PBS) without feeder cells in Dulbecco’s Modified Eagle Medium (DMEM) (high glucose, 4.5 g/L) supplemented with 15% (v/v) European fetal calf serum (EFCS), 2 mM nonessential amino acids (NEAA), 100 U/mL penicillin, 100 µg/mL streptomycin, 0.1 mM β-mercaptoethanol, and 1,000 U/mL Leukaemia Inhibitory Factor (LIF) in a humidified atmosphere containing 5% CO2/air at 37°C. Cells were quiesced in DMEM containing 1% (v/v) EFCS, 2 mM NEAA, 100 U/mL penicillin, 100 µg/mL streptomycin, 0.1 mM β-mercaptoethanol, and 1,000 U/mL LIF prior to stimulation with agonists.
CGR8 cells were cultured on 0.2% (w/v) gelatin/PBS without feeder cells in Glasgow Minimum Essential Medium (GMEM) supplemented with 10% (v/v) EFCS, 2 mM NEAA, 100 U/mL penicillin, 100 µg/mL streptomycin, 0.5 mM β-mercaptoethanol, and 100 U/mL LIF in a humidified atmosphere containing 5% CO2/air at 37°C. Cells were quiesced in DMEM containing 1% (v/v) EFCS, 2 mM NEAA, 100 U/mL penicillin, 100 µg/mL streptomycin, 0.1 mM β-mercaptoethanol, and 100 U/mL LIF prior to agonist stimulation.
Chinese hamster ovary (CHO) cells were cultured in DMEM supplemented with 10% (w/v) EFCS, 100 U/mL penicillin, and 100 µg/mL streptomycin in a humidified atmosphere containing 5% CO2 at 37°C. MDA-MB-453 breast cancer cells were obtained from ATCC (Rockville, MD) and were grown in a monolayer culture in high glucose DMEM with 10% EFCS, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37°C with 5% CO2. Airway smooth muscle (ASM) cells were maintained in DMEM supplemented with 10% (v/v) fetal calf serum and 10% (v/v) horse serum. Cells were routinely used at passage 3–4.
S1P receptor transfection
Chinese hamster ovary cells were transfected with HA-S1P5 plasmid construct (1 µg per well) using the Lipofectamine™ 2000 reagent (Invitrogen, UK) according to the manufacturer’s instruction. Tranfection was performed for 24 h at 37°C before serum starvation for a further 24 h prior to cell stimulation.
Reverse transcriptase-polymerase chain reaction (RT-PCR)
RNA was extracted from ES-D3 cells and CGR8 cells. Proliferating ES-D3 and CGR8 cells were grown to ∼80% confluence in gelatin-coated 25 cm2 flasks for RNA extraction using the Nucleospin II RNA Extraction kit (Abgene). To eliminate any remaining residual genomic DNA, total RNA was subjected to further DNase I treatment. To 1 µg total RNA, 1 µL of 10× DNase buffer and 1 µL of DNase I were added in a total volume of 10 µL and incubated at room temperature for 15 min. The reaction was terminated by adding 1 µL of 25 mM EDTA and incubated at 65°C for 10 min, followed by 1 min on ice. Total RNA was stored at −70°C.
Reverse transcription was performed using Superscript II (Invitrogen, UK). Five micrograms of DNase I-treated total RNA was combined with 0.5 µg oligo(dT)12–18, 0.5 µL of 20 mM dNTP mixture, and RNAse/DNase-free water to 12 µL final volume, then heated to 65°C for 5 min. Following a quick chill on ice, 4 µL of 5× First Strand buffer (50 mM Tris-HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2) and 2 µL of 0.1 M DTT were added and incubated at 42°C for 2 min. After that, 1 µL of Superscript II RT (200 units) was added and the reaction heated to 42°C for 50 min, followed by 15 min at 70°C, to inactivate the reaction. For each first strand reaction, a separate reaction excluding Superscript II RT was performed to ensure there was no genomic contamination. For these reactions (-RT), Superscript II RT was replaced by 1 µL of distilled water (RNase/DNase-free). The cDNA produced was stored at −20°C for use as a DNA template in polymerase chain reaction (PCR).
PCR was performed using Taq DNA polymerase in a 50 µL reaction containing 5 µL of 10× reaction buffer (containing 200 mM Tris-HCl (pH 8.4) and 500 mM KCl), 1.5 µL of 50 mM MgCl2, 1 µL of 20 mM dNTP mix, 1 µL each of forward and reverse primer (50 pmol/mL), 2 µL of template DNA, 38.6 µL RNAse/DNase-free water, and 0.4 µL Taq polymerase (5 U/µL). For each PCR, a negative control reaction (blank) was included where cDNA template was omitted. Reactions were performed in a Phoenix thermal cycler using conditions optimized for each primer pair. A typical reaction was: initial denaturation of the template at 94°C for 5 min, 35 cycles of 94°C for 30 s (denaturation), 30 s at the specific annealing temperature for each primer set, 72°C for 1 min (extension), followed by final primer extension at 72°C for 10 min. Amplification products were analyzed by agarose gel electrophoresis and their identity confirmed by sequencing. The primers are listed in Table 1.
P
Western blot analysis
Analysis of proteins by SDS-PAGE and Western blot analysis was performed as described previously by us [16].
DNA synthesis
ES-D3 cells and CGR8 cells were seeded onto 24-well plates in 0.5 mL of their normal culture media and grown to 60%–70% confluence. The medium was replaced with medium supplemented with 1% (v/v) EFCS, 2 mM NEAA, penicillin, streptomycin, and LIF for 24 h. S1P receptor antagonists/inverse agonists were added to cells for 8.5 h. [3H]Thymidine (37 KBq per well) was added for a further 60 min (9.5 h total). The medium was aspirated and cells were washed three times for 10 min with 1 mL of 10% (w/v) ice-cold trichloroacetic acid. Nuclear material was dissolved in 0.25 mL of 0.1% SDS/0.3 M NaOH and transferred to scintillation vials with 2 mL of scintillant, and the incorporation of [3H]thymidine into newly synthesized DNA was quantified by liquid scintillation counting.
Quantification
Densitometric quantification of Western blots was achieved using Scion program software. Phosphorylated ERK-2/total ERK-2 ratios were calculated and expressed relative to unstimulated control cells (100%).
Results and Discussion
ES-D3 and CGR8 cells express S1P receptors
RT-PCR analysis using gene-specific primers demonstrated that both ES-D3 and CGR8 cells express S1P1, S1P2, and S1P3 mRNA transcript (confirmed by nucleotide sequencing) (Fig. 1A and 1B). The S1P4-specific primers generated an amplicon of the predicted size from a S1P4 plasmid construct, used as a positive control. No amplicon of this size was amplified from ES-D3 or CGR8 cells, indicating the absence of S1P4 mRNA transcript in these cells. A nonspecific amplicon (∼600 bp) was generated using mRNA from CGR8 cells (Fig. 1C). Sequencing identified this to be nucleophosmin 1 (data not shown). In addition, Western blot analysis of ES-D3 cell lysates with anti-S1P5 antibody immunostained a single protein with a Mr = 42 kDa, which corresponds with the native molecular mass of S1P5 (Fig. 1D). Similarly, a 42 kDa protein was the major immunoreactive protein detected in CGR8 cell lysates with anti-S1P5 antibody. These findings contrast with studies from Kleger and colleagues [17], who demonstrated the presence of all five S1P receptor subtypes in R1 mouse ES cells. These cells differ from ES-D3 and CGR8 ES cells in that each is derived from different mice strains (R1 is from 129X1 × 129S1 crossed mice; ES-D3 from 129S2/SvPas mice and CGR8 from 129 mice) and suggests different phenotypic expression profiles.

Expression of sphingosine 1-phosphate (S1P) receptors in embryonic stem (ES)-D3 and CGR8 cells. RT-PCR analysis with gene-specific primers showing expression of S1P1, S1P2, and S1P3 receptor mRNA transcript in (
S1P induces activation of ERK-1/2
We next investigated whether S1P regulates the ERK-1/2 pathway in ES cells. An S1P receptor-mediated response is implicated by findings that pretreatment of ES-D3 or CGR8 cells with 0.5 µg/mL pertussis toxin (PTX, which functions to catalyze ADP-ribosylation of C-terminal cysteine in Giα and thereby uncouples heterotrimeric Gi from the GPCR) reduced the S1P-dependent activation of ERK-1/2 (Fig. 2A). Therefore, the S1P receptor involved appears to be coupled to the G-protein, Gi. To further explore the identity of the S1P receptor involved, we used a pharmacological approach with compounds that demonstrate selectivity at different S1P receptors, functioning either as inverse agonists or as competitive antagonists. In this regard, we found that treatment of either ES-D3 or CGR8 cells with SB649146 [16], JTE013 [18], or CAY10444 [19] had no effect on the S1P-induced activation of ERK-1/2 (Fig. 2B–2D). We confirmed that these compounds were effective at reducing S1P signaling in cells known to express respective S1P receptors. Thus, the pretreatment of ASM cells (which we have previously shown is mediated by S1P1 [16,20]) with SB649146 reduced S1P-stimulated activation of ERK-1/2 (Fig. 2E). JTE013 reduced activation of ERK-1/2 by S1P (and was without effect on EGF) in MDA-MB-453 cells, which express S1P2 (Fig. 2E).

Sphingosine 1-phosphate (S1P) regulation of ERK-1/2 signaling in embryonic stem (ES)-D3 and CGR8 cells. ES-D3 and CGR8 cells were pretreated with either pertussis toxin (PTX; 0.1 or 0.5 µg/mL) for 24 h or SB649146 (1 or 10 µM), JTE013 (1 or 10 µM), or CAY10444 (1 or 10 µM) for 15 min and then treated with or without S1P (5 µM) for 10 min. The Western blot shows the (
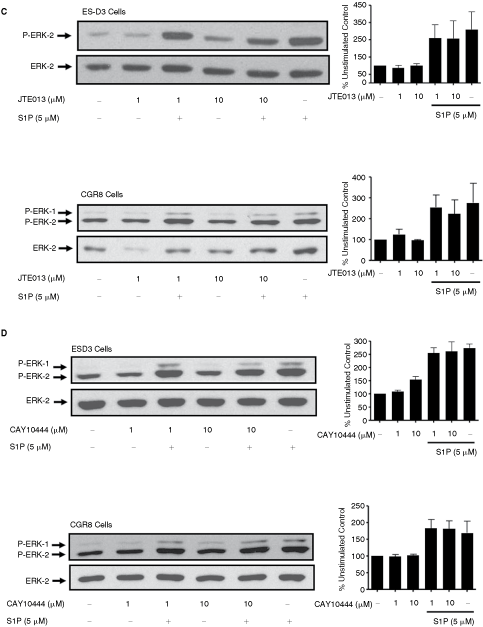
Continued.

Continued.
Two additional lines of evidence were obtained to indicate that the S1P1 is not involved in regulating the ERK-1/2 pathway in ES cells. We and others have shown that S1P1 functionally interacts with PDGFβ receptor to regulate ERK-1/2 and cell migration in several mammalian somatic cells [21–23]. However, we found that PDGF was ineffective in stimulating ERK-1/2 activation in ES-D3 and CGR8 cells and combined stimulation of these cells with S1P/PDGF did not demonstrate synergistic activation of ERK-1/2 (Fig. 3). These results contrast with the synergistic interaction between PDGF and S1P in terms of the stimulation of ES differentiation [13]. Our findings suggest a lack of cross-regulation between S1P and PDGF and therefore functional S1P1 in terms of the regulation of the ERK-1/2 pathway. Moreover, we found that the stimulation of ES-D3 cells with SEW2871, an S1P1 selective agonist [24], was without effect on the ERK-1/2 pathway (Fig. 3), thereby excluding a functional role for S1P1 in regulating ERK-1/2.
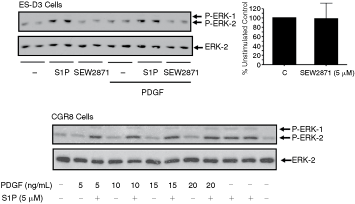
Sphingosine 1-phosphate (S1P) and platelet-derived growth factor (PDGF) regulation of ERK-1/2 signaling in embryonic stem (ES)-D3 and CGR8 cells. ES-D3 and CGR8 cells were treated with and without S1P (5 µM), PDGF (10 ng/mL, ES-D3 cells; 5–20 ng/mL, CGR8 cells) or SEW2871 (5 µM) for 10 min. The Western blot shows the lack of synergistic interaction between S1P and PDGF in terms of ERK-1/2 activation in CGR8 and ES-D3 cells and the lack of effect of SEW2871 on ERK-1/2 activation in ES-D3 cells. The histogram represents densitometric quantification of P-ERK-2/ERK-2 ratios for n = 3 experiments showing no effect of SEW2871 on ERK-1/2 activation. Phosphorylated ERK-1/2 was detected on Western blots probed with anti-phospho ERK-1/2 antibody. Blots were also probed with anti-ERK-1/2 antibody to ensure equal protein loading.
DNA synthesis in ES-D3 and CGR8 cells
We next investigated the effect of S1P receptor antagonists on DNA synthesis, which was used as a marker for proliferation. We used SB649146, JTE013, or CAY10444 to establish whether S1P1, S1P2, or S1P3 are involved in regulating basal DNA synthesis. In this regard, SB649146 and JTE013 but not CAY10044 reduced basal DNA synthesis (Fig. 4A–4C), suggesting that S1P1 and S1P2 but not S1P3 might be involved in regulating basal DNA synthesis.
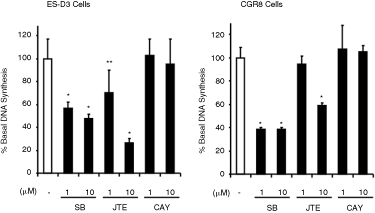
DNA synthesis. Embryonic stem (ES)-D3 and CGR8 cells were treated with either SB649146 (1 and 10 µM), JTE013 (1 and 10 µM), or CAY10444 (1 and 10 µM) for 9.5 h. The histograms show the effect of SB649146, JTE013, CAY1044 on basal DNA synthesis, measured as [3H]thymidine incorporation (% inhibition of basal DNA synthesis, n = 4–12). *P < 0.01, **P < 0.05 (unpaired t-test vs. control).
Phyto-S1P and S1P5
Despite evidence to suggest a lack of a role for S1P1–3 (and the absence of S1P4 expression) in regulating ERK-1/2, the PTX sensitivity of S1P signaling implicates the involvement of a GPCR. Therefore, we considered a possible role for the S1P5 receptor, which is expressed in both ES cell types (Fig. 1D). We therefore looked for compounds that bind to S1P5 and that could be used as specific tools to interrogate the function of the S1P5 receptor in ES cells. Phytosphingosine 1-phosphate (phyto-S1P) and diastereomeric DM95 and DM97 (Fig. 5A) were synthesized from suitable precursors. Phyto-S1P has been identified as being an agonist of S1P4, with no activity at S1P1, S1P2, or S1P3 receptors [25,26]. We therefore tested whether these compounds have agonist activity at the S1P5 receptor. To achieve this we transfected CHO cells with HA-tagged S1P5 receptor plasmid construct, which results in expression of the receptor in CHO cells (Fig. 5B). We found that phyto-S1P, DM95, and DM97 induced activation of ERK-1/2 in CHO cells overexpressing recombinant S1P5 (Fig. 5B). We therefore treated ES-D3 and CGR8 cells with phyto-S1P, DM95, or DM97 to interrogate possible S1P5 function, and showed that these compounds were able to activate ERK-1/2 (Fig. 5C).
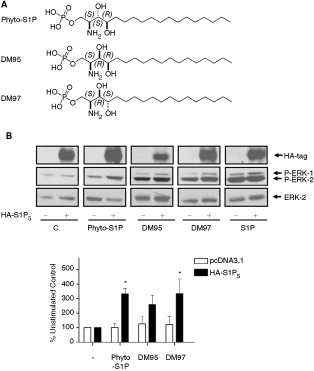
Effect of sphingosine 1-phosphate (S1P) and phyto-S1P on ERK-1/2 activation in Chinese hamster ovary (CHO) cells overexpressing recombinant S1P5 and embryonic stem (ES) cells. (

Continued.
S1P-dependent activation of ERK-1/2 involves PKC and c-Src
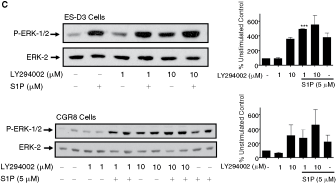
We next investigated the mechanism by which S1P activates the ERK-1/2 pathway in ES-D3 and CGR8 cells. Previous studies have demonstrated the involvement of c-Src, PKC, and PI3K in regulating S1P receptor signaling in somatic cells [27]. Pretreatment of both ES-D3 and CGR8 cells with PP2 (c-Src inhibitor) or bisindolylmaleimide (PKC inhibitor) reduced the activation of ERK-1/2 by S1P (Fig. 6A, 6B). In contrast, treatment of cells with LY29004 (PI3K inhibitor) had no inhibitory effect on S1P-stimulated ERK-1/2 activation and on its own stimulated ERK-1/2 (Fig. 6C).
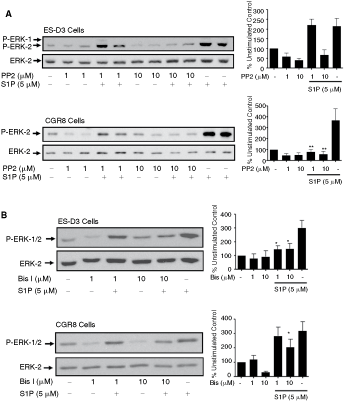
Sphingosine 1-phosphate (S1P) regulation of ERK-1/2 signaling in embryonic stem (ES)-D3 and CGR8 cells via c-Src and PKC. ES-D3 and CGR8 cells were pretreated with PP2 (1 and 10 µM), bisindolylmaleimide I (1 and 10 µM), or LY294002 (1 and 10 µM) for 15 min and then treated with or without S1P (5 µM) for 10 min. Western blots show the effect of (
Continued.
Conclusion
Our findings suggest that S1P may induce activation of ERK-1/2 via an S1P5 receptor-dependent mechanism in mouse ES cells, thereby highlighting a novel role for this receptor in regulating this kinase pathway module. In human ES cells, ERK-1/2 appears to protect against apoptosis [15]. It remains to be determined whether ERK-1/2 has a similar role in mouse ES cells. However, if this is the case, then our data might suggest that S1P5-mediated activation of ERK-1/2 represents an antiapoptotic/cell survival signaling program. In addition, our findings with SB649146 and JTE013 suggest a role for S1P1/S1P2 in regulating proliferation that is independent of ERK-1/2 signaling. Finally, our data indicate that phyto-S1P might be a useful pharmacological reagent for probing S1P5 function in ES or somatic cells that lack S1P4.
Footnotes
Acknowledgments
This work was supported by BBSRC funding to S.P. and N.J.P. D.M. acknowledges the former Ministerio de Ciencia y Tecnologia (Spain) for a predoctoral fellowship and a scholarship.
Author Disclosure Statement
None of the authors have a competing financial interest.