
Abstract
Embryonic stem cells (ESCs) proliferate rapidly and have a unique cell-cycle structure with a very short G1 phase. Previous reports suggested that the rapid G1 phase progression of ESCs might be underpinned by high and precocious Cdk2 activity and that Cdk2 activity might be crucial for both cell-cycle regulation and cell-fate decisions in human ESCs. However, the actual role of Cdk2 in cell-cycle progression of mouse ESCs (mESCs) has not been elucidated. In this study, we investigated the effects of down-regulation of Cdk2 activity by olomoucine II in 2 mESC lines. Olomoucine II treatment significantly increased the G1 phase cell numbers, decreased the S phase cell numbers, and inhibited DNA replication in mESCs. In nocodazole-synchronized mESCs, we show that specific down-regulation of Cdk2 activity prolongs G1 phase progression. In addition, down-regulation of Cdk2 activity in mESCs established a somatic cell-like cell cycle and induced expression of differentiation markers. Our results suggest that high Cdk2 activity is essential for rapid G1 phase progression and establishment of ESC-specific cell-cycle structure in mESCs and support the hypothesis of a link between cell-cycle regulation and pluripotency maintenance in ESCs. This study reveals olomoucine II to be an effective tool for manipulation of the cell cycle and pluripotency in ESCs and very likely also for the manipulation of other stem cell types, including cancer stem cells.
Introduction
E
At the molecular level, the rapid cell-cycle progression of ESCs in vitro has been described as the result of an unusually high and precocious Cdk2 activity in mouse ESCs (mESCs) that appears to be cell-cycle-independent [3]. In contrast, Cdk2 activity in somatic cells is tightly regulated as Cdk2 is involved in both G1/S transition and initiation of DNA replication [10,11], processes that require proper timing and cellular conditions to ensure flawless genome replication and production of viable progeny [12]. Proliferation in vivo in early embryogenesis appears to be independent of Cdk2 [13,14], suggesting that the G1/S phase-associated Cdks can substitute for each other [15 –17]. The study by Stead et al. [3] suggested that the absolute level of Cdk2 activity in mESCs determines cell division rates. A recent study in human ESCs (hESCs) suggested that Cdk2 activity might be crucial for both cell-cycle regulation and cell-fate decisions because siRNA knockdown of Cdk2 resulted in G1 phase arrest and differentiation of hESCs to extraembryonic lineages [18]. But the actual role of Cdk2 in cell-cycle progression of mESCs is not fully understood, and, moreover, Cdk2 is predominantly localized on centrosomes in mESCs (our observation, data not shown), which makes its role in mESC cycle regulation even more obscure.
Selective chemical Cdk inhibition is a useful tool for deciphering the roles and requirements in cells of individual Cdks [19]. In our study, we used this approach to investigate the role of Cdk2 in G1 phase progression of mESCs. We took advantage of 2 new generation Cdk inhibitors: olomoucine II and CAN508. We used olomoucine II to inhibit Cdk2 activity in mESCs. As olomoucine II is a potent inhibitor of both Cdk2 and Cdk9 [20], we used also CAN508, a specific Cdk9 inhibitor [21], to distinguish the effects of Cdk2 inhibition from those of Cdk9 inhibition. Cdk2 inhibition by olomoucine II treatment efficiently increases the proportion of G1 cells in mESC culture. In nocodazole-synchronized cells, we show that this is due to decelerated progression of mESCs through the G1 phase. Prolonged down-regulation of Cdk2 activity established a somatic cell-like cell cycle and induced morphology and gene expression changes indicative of mESC differentiation. Based on kinase assays, on comparison of the results of CAN508 and olomoucine II treatment in mESCs, and on the effects of Cdk2 knockdown by siRNA, we suggest that the observed cell-cycle structure changes and induction of differentiation in mESCs is the result of down-regulation of Cdk2 activity after olomoucine II treatment. Our results suggest that the high Cdk2 activity plays an important role in the regulation of rapid G1 phase progression as well as in the establishment of the ESC-specific cell-cycle structure and maintenance of pluripotency in mESCs.
Materials and Methods
Cell culture and synchronization
For our studies, 2 different mESC lines were used; an inbred HM1 cell line derived from 129 mouse strain [22] and an F1 (129SvJae×C57BL/6) hybrid line V6.5 (Open Biosystems, Huntsville, AL). The total in vitro culture time of these mESCs at the time of experimental testing was equivalent to passage number 22–35 of the original mESC line. Both mESC lines were checked repeatedly for chromosome number and possible karyotypic abnormalities. mESC culture was carried out following standard procedures [23]. In brief, the cells were maintained on culture dishes covered with 0.1% gelatin (Sigma-Aldrich, St. Louis, MO) in a humidified atmosphere of 10% CO2 at 37°C in high-glucose Dulbecco’s modified Eagle’s medium (D-MEM) with GlutaMAX (Invitrogen/Gibco, Grand Island, NY) supplemented with 15% fetal bovine serum (FBS; Thermo Fisher Scientific/HyClone, Waltham, MA), 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 2 mM glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin (Gibco), 0.1 µM β-mercaptoethanol (Serva), and 1000 U/mL leukemia inhibitory factor (ESGRO; Chemicon, Temecula, CA).
Human colon adenocarcinoma cell line HT29 (ATCC) was maintained in a humidified atmosphere of 10% CO2 at 37°C in high-glucose D-MEM with GlutaMAX supplemented with 10% FBS (Gibco), 100 U/mL penicillin, and 100 µg/mL streptomycin.
mESCs were synchronized in G2/M phase by 400 nM nocodazole (Sigma-Aldrich) treatment for 12 h. After mitotic shake-off, cells were washed 3 times with phosphate-buffered saline (PBS) with 1% FBS (Gibco) and plated in standard mESC media.
Inhibitors
Olomoucine II (2-{[2-((1-R)-1-hydroxymethyl-propylamino)-9-isopropyl-9Hpurin-6-ylamino]-methyl}-phenol) and CAN508 (4-[(3,5-diamino-1H-pyrazol-4-yl)diazenyl]phenol) were synthesized according to published procedures [21,24]. Both olomoucine II and CAN508 were stored as 100 mM stock solutions in DMSO.
MTT assay
Equal numbers of cells per well (7,000 for mESCs, 5,000 for HT29) were plated on a 96-well plate and incubated for 3 h (mESCs) or 24 h (HT29) under standard conditions. Triplicate samples of these cells were treated with increasing concentrations of inhibitors (range from 10−8 to 10−4 M) or mock-treated and incubated for 24 h (mESCs) or 72 h (HT29) under standard conditions. After this time, MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide; Sigma-Aldrich) was added to a final concentration 0.5 mg/mL and the cells were incubated for another 2.5 h under standard conditions. The media was then removed, 100 µL of 10% sodium dodecyl sulfate (SDS) was added per well, and the 96-well plate was incubated overnight at room temperature on a shaker. The absorbance was read at 570 nm. To calculate IC50 values (concentrations that produce a 50% of inhibitory effect on cell proliferation), the results from all triplicates were transformed to percentage of controls, and plotted as sigmoid dose–effect curves using a nonlinear regression mode and the GraphPad Prism 5 software. Using this software, the IC50 values were interpolated.
Flow cytometric analysis
Cell-cycle distribution was evaluated by 5-bromo-2′-deoxyuridine (BrdU) incorporation and propidium iodide staining. Cells were pulsed with 10 µM BrdU (Sigma-Aldrich) for 30 min, trypsinized (0.5% trypsin–ethylenediaminetetraacetic acid [EDTA]; Gibco) to obtain a single cell suspension, washed twice in PBS with 1% FBS, and resuspended in PBS. The cells were then fixed in ice cold 70% ethanol. After rehydration in PBS with 1% FBS (Gibco), cells were incubated in 2 M HCl with 0.5% (v/v) Triton X-100 for 30 min at room temperature. Following neutralization with 0.1 M Na2B4O7, cells were collected by centrifugation and washed with PBS with 1% FBS and 0.5% (v/v) Tween-20. Then they were stained with anti-BrdU fluorescein isothiocyanate (FITC)-labeled antibody (1:20; Roche Diagnostics, Mannheim, Germany) for 30 min at room temperature in darkness. The cells were then washed with PBS with 1% FBS and 0.5% (v/v) Tween-20 and incubated in 1.1% sodium citrate dehydrate with 5 ng/µL ribonuclease A (DNA Lego Ribonuclease A; Top-Bio, Prague, Czech Republic) and 60 µg/µL propidium iodide (Sigma-Aldrich) for 30 min at 37°C in the dark. Cells were analyzed by flow cytometry on the Cytomics FC 500 machine using the CXP software (Beckman Coulter, Fullerton, CA) and following standard procedures. Analysis by MultiCycle (Phoenix Flow Systems) was applied to assess cell-cycle distribution.
Western blot analysis
Cells on culture dishes were washed in cold PBS, collected into IP buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM EDTA, 2.5 mM ethyleneglycoltetraacetic acid (EGTA), 10% (v/v) glycerol, 0.1% (v/v) Tween-20, 1 µM dithiothreitol (DTT), 1 µM NaF, 10 µM β-glycerophosphate, 10 µg/mL leupeptin, 2 µg/mL aprotinin, 0.1 µM Na3VO4, 0.1 µM PMSF) and incubated for 1 h at 4°C. Lysates were cleared by centrifugation at 18,800g at 4°C for 30 min. Proteins were electrophoretically resolved on SDS-polyacrylamide gels and electroblotted onto nitrocellulose membranes (Bio-Rad). Membranes were incubated at 4°C overnight with primary antibodies [anti-α-tubulin (Sigma-Aldrich); Cdk2 (M2) (Santa Cruz Biotechnology, Santa Cruz, CA); RNA polymerase II antibody [H5], RNA Polymerase II antibody [8WG16] (Abcam)] at dilution recommended by the supplier, washed in PBS with 0.05% Tween, and incubated for 1 h with anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibody (DakoCytomation, Glostrup, Denmark). HRP activity was detected with an ECL detection kit (Pierce) on films (Bio-Rad, Hercules, CA).
Immunoprecipitation and kinase assay
Immunoprecipitations were conducted at 4°C. Protein A Agarose Beads (Sigma-Aldrich) were incubated for 1 h on a rotor in 1 mL IP buffer with 5 µL Cdk2 antibody (Cdk2 (M2); Santa Cruz Biotechnology) or 4 µL Cdk1 antibody (Anti-cdk1/cdc2, CT; Upstate Biotechnology, Lake Placid, NY). After washing 3 times with IP buffer, the agarose beads were incubated for 1 h with protein lysates (200 µg of proteins) in IP buffer on a rotor and finally washed 3 times with IP buffer. Agarose beads with immunocomplexes were equalized with kinase assay buffer (KAB; 50 mM HEPES pH 7.5, 10 mM MgCl2, 5 mM MnCl2, 2.5 mM EGTA, 100 µM β-glycerophosphate, 2 µM NaF, 1 µM DTT, 0.1 µM Na3VO4). They were then resuspended in 30 µL kinase reaction mixture (18 µL KAB, 9 µL 75 µM adenosine triphosphate (ATP) in KAB, 2 µg histone H1, 1 µL [γ-33P]ATP (10 µCi; MP Biochemicals, Irvine, CA) and incubated for 30 min at 30°C. Reactions were terminated by addition of 12 µL of 4× Loading sample buffer (8% SDS, 40% glycerol, 400 mM DTT, 240 mM Tris, pH 6.8, 0.004% bromophenol blue). Samples were boiled and electrophoretically separated on 8%–12% SDS-polyacrylamide gels. Radioactivity of the dried gels was detected with the bioimager BAS 1800 (Fuji Photo Film Co., Ltd.).
Statistical analysis
Statistical evaluation of the data was performed with the independent 2-sample t-test using Statistica 8.0. P < 0.05 was considered statistically significant.
RNA isolation and quantitative real-time RT-PCR
Total RNA from cells was isolated using TRI Reagent (Ambion) and treated with DNAse (TURBO DNA-free, Ambion). One microgram of total RNA was used for reverse transcription by First Strand cDNA Transcriptor Synthesis Kit (Roche Applied Science) according to the manufacturer’s instructions. The relative quantity of target genes’ RNA was determined by real-time quantitative polymerase chain reaction (PCR) using a LightCycler 480 (Roche). Gene-specific primer pairs (listed in Supplementary Table 1; Supplementary materials are available online at http://www.liebertpub.com/) were designed and evaluated at an annealing temperature of 60°C using freely available Web-based software in the Universal ProbeLibrary Assay Design Center. The amplification mix and program were prepared using LightCycler 480 SYBR Green I Master (Roche) according to the manufacturer’s instructions. Data were analyzed by using LightCycler 480 Relative Quantification Software (Roche) and expression levels were normalized to the housekeeping gene GAPDH.
IC50 V
The number of viable cells after 24 h (V6.5 and HM1 cells) or 72 h (HT29 cells) treatment with olomoucine II or CAN508 was quantified in MTT assays and IC50 values were calculated from dose–response curves. The results are means (±SD) of at least 3 experiments.
siRNA transfection
mESCs were transfected with 70 nM Stealth Select RNAi for mouse CDK2 (Invitrogen, Carlsbad, CA; sequence of the duplex: 5′-CCCUUUCUUCCAGGAUGUAACUAAA-3′) or 70 nM Stealth RNAi Negative Control Duplex with Medium GC content (Invitrogen) using X-tremeGENE siRNA Transfection Reagent (Roche) as outlined in the manufacturer’s instructions. The cells were analyzed 24 h after transfection.
Quantification of Cdk2 protein levels
Cdk2 protein levels were quantified from scanned images of western blot analyses using AlphaEase FC software, Version 5.0.1 (Alpha Innotech, San Leandro, CA).
Results
Cdk inhibitors are cytotoxic for mESCs
The cytotoxicity of the Cdk inhibitors olomoucine II and CAN508 was tested in MTT assays for 2 mESC lines, V6.5 and HM1, and for the adenocarcinoma cell line HT29. HT29 cells were used as a reference cell line because their response to olomoucine II and CAN508 treatment has been described already [20,21]. mESCs were treated for 24 h and HT29 for 72 h with scaled doses of each inhibitor separately. The duration of inhibitor treatment corresponded approximately to 3 cell cycles in the respective cell lines. The results of MTT tests are summarized in Table 1. The cytotoxic potential of the pan-specific inhibitor olomoucine II was stronger than that of the cytotoxic potential of Cdk9-specific inhibitor CAN508. IC50 of olomoucine II was 9.0 µM for V6.5, 6.3 µM for HM1, and 10.7 µM for HT29. The HT29 IC50 value is consistent with the observation of Krystof et al. [20]. IC50 for CAN508 was 129.0 µM for V6.5, 68.1 µM for HM1, and 54.4 µM for HT29 (this value is consistent with Krystof et al. [21]).
Treatment of mESCs with Cdk inhibitors causes major changes in cell-cycle progression
To investigate the effects of olomoucine II and CAN508 on the cell cycle, mESCs and HT29 cells were treated with olomoucine II or CAN508 at IC50 concentrations for the time approximately corresponding to the duration of 1 cell cycle (11 h for mESCs, 24 h for HT29). To investigate more immediate effects of Cdk inhibition, cells of all 3 lines were incubated with the inhibitors for 3 h only. Following incubation, cells were double-stained with BrdU and propidium iodide and their cell-cycle profiles were analyzed by flow cytometry. The results for 1 experiment, representative of 3 independent experiments, are depicted in Figure 1. In the control cell line HT29, the 24-h treatment with olomoucine II caused a slight, but statistically significant increase (from 72% in mock-treated cells to 75% in olomoucine II-treated cells; P = 0.003) in the proportion of G1 cells and a minor decrease (P = 0.06) in the proportion of S phase cells (Fig. 1). However, the 24-h treatment with CAN508, a specific inhibitor of transcriptional kinase Cdk9, led to a more prominent decrease in G1 phase cell number (from 72% in mock-treated cells to 63% in CAN508-treated cells; P = 0.009) and a prominent increase (from 22% in mock-treated cells to 35% in CAN508-treated cells; P = 0.02) in S phase cell number (Fig. 1). The same trend to the cell-cycle changes was observed as early as 3 h of inhibitor treatment (Fig. 1). Neither olomoucine II nor CAN508 treatment of HT29 cells led to inhibition of DNA replication (Fig. 1).
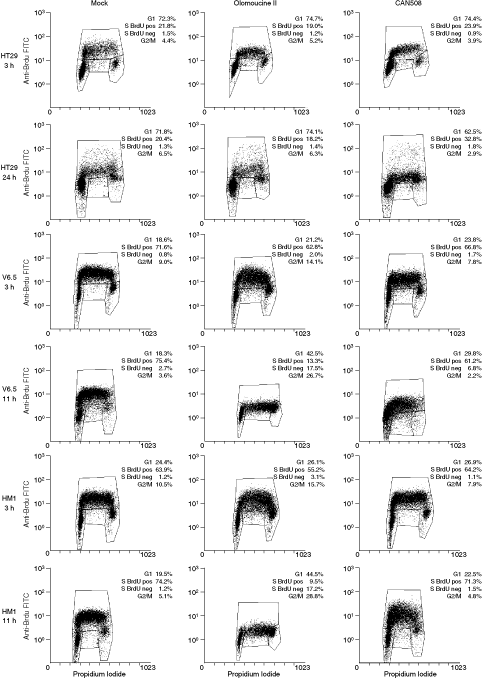
Changes of cell-cycle profiles after Cdk inhibitor treatment. HT29, V6.5, and HM1 cells were treated with olomoucine II or CAN508 (at the concentrations corresponding to their IC50 values) or mock-treated for the indicated times. Cells were subsequently pulsed with 5-bromo-2′-deoxyuridine (BrdU) for 30 min, fixed, stained with propidium iodide and their cell-cycle profile was analyzed by flow cytometry. The figures are representatives of 3 independent experiments.
mESCs responded to the inhibitor treatment differently from the HT29 cells, and similarly to each other. An 11-h treatment with olomoucine II caused a significant increase in G1 phase cell number in mESCs: from 18% to 43% in mock-treated V6.5 cells (P = 0.01; Fig. 1) and from 20% to 45% in mock-treated HM1 cells (P = 0.04; Fig. 1). Correspondingly, the proportion of mESCs in S phase dropped significantly after 11-h olomoucine II treatment (from 78% to 31% in V6.5 cells and from 75% to 27% in HM1 cells; P = 0.03 and 0.05, respectively). The proportion of G2 phase cells exhibited changes as well, but these changes were not statistically significant (P = 0.09 for V6.5 cells and 0.06 for HM1 cells). Moreover, 11-h treatment with olomoucine II caused a 65-fold and a 137-fold increase of BrdU-negative cells in V6.5 and HM1 mESCs, respectively, pointing to a significant inhibition of DNA replication (P = 0.02 and 0.01, respectively).
Treatment with the Cdk9 inhibitor CAN508 had similar, but less prominent consequences to those of olomoucine II treatment: G1 phase cell numbers increased (P = 0.006 for V6.5 and P = 0.37 for HM1), S phase cell numbers decreased (P = 0.03 for V6.5 and P = 0.06 for HM1), and the proportion of G2 phase cells decreased (P = 0.28 and 0.36 for V6.5 and HM1, respectively). The number of BrdU-negative S phase cells increased (Fig. 1), but these slight changes were not statistically significant (P = 0.18 and 0.15 for HM1 and V6.5 cells, respectively). Again, the propensity for these cell-cycle changes (observed after 11-h treatment with inhibitor) can be observed as early as 3 h after inhibitor treatment in mESCs.
Effects of CAN508 and olomoucine II treatment on Cdk9, Cdk2, and Cdk1 activities in mESCs
Olomoucine II and CAN508 were reported to be effective inhibitors of Cdk9 both in in vitro kinase inhibition assays and in cancer cell lines: IC50 for Cdk9–cyclin T is 0.06 µM for olomoucine II [20] and 0.35 µM for CAN508 [21]. To check the effectiveness of Cdk9 inhibition by CAN508 or olomoucine II, we tested, using western blots, the phosphorylation status of RNA polymerase II on Ser2 in the C-terminal domain (RNA-pol II CTD), the specific Cdk9 target site. We found that CAN508 effectively abolishes Cdk9 activity at the concentrations used in our experiments, as it down-regulates the phosphorylation of RNA-pol II CTD at a concentration of 100 µM in both studied ESC lines (Fig. 2). Cdk9 activity was also inhibited by olomoucine II in mESCs, where a decrease of CTD phosphorylation was detected at 2.5 µM olomoucine II in V6.5 cells and at 5 µM olomoucine II in HM1 cells (Fig. 2).
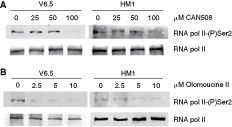
Phosphorylation of the C-terminal domain of RNA polymerase II is abolished by CAN508 and olomoucine II treatment. Cells were treated for 11 h with the indicated concentration of CAN508 (
Besides Cdk9, olomoucine II exhibits specificity for Cdk2 (IC50 for Cdk2–cyclin E is 0.1 µM [20]) and a lower specificity for Cdk1 (IC50 for Cdk1–cyclin B is 7.6 µM [20]). To determine the activity of olomoucine II against Cdk2 and Cdk1 in mESCs, we undertook a series of histone H1 kinase assays for Cdk2 and Cdk1 activities in mock- or olomoucine II-treated mESCs. The results are plotted in Figure 3. Following 5 µM olomoucine II treatment, the activity of Cdk2 significantly decreased (to 42% of the Cdk2 activity in mock-treated cells; P = 0.002). In contrast, the activity of Cdk1 was not significantly decreased by up to 10 µM olomoucine II (64% of the Cdk1 activity in mock-treated cells; P = 0.15).
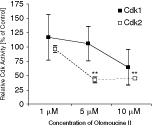
Inhibition of Cdk1 and Cdk2 activity by olomoucine II treatment. After 1-h treatment of V6.5 cells with designated concentrations of olomoucine II, Cdk1 or Cdk2 activity was measured in histone H1 kinase assays. The results were normalized to control (mock-treated cells). The data are the means of 2 independent experiments. Statistical significance was assessed using Student’s t-test, **P < 0.005. Bars = 95% confidence interval.
CAN508 was reported to have minor inhibitory effect on Cdk2 (IC50 for Cdk2–cyclin E is 3.5 µM [21]) in somatic cells. The specificity of Cdk9 inhibition by CAN508 in mESCs was checked in kinase assays for Cdk1 and Cdk2 activities. CAN508 treatment (up to IC50 concentration for the respective mESC lines) had no effect on Cdk1/Cdk2 activity (data not shown).
These observations suggest the Cdk inhibitors olomoucine II and CAN508 have the same targets in mESCs and in somatic cells.
Olomoucine II treatment blocks DNA replication in mESCs
From kinase assays, we concluded that 5 µM is the upper concentration limit of olomoucine II that effectively inhibits Cdk2 activity while not affecting Cdk1 activity (Fig. 3). This results suggested use of 5 µM olomoucine II for more specific down-regulation of Cdk2 activity in mESCs. In view of this observation, both V6.5 and HM1 cells were treated with 1, 2.5, or 5 µM olomoucine II, or mock-treated to investigate the effects of down-regulation of Cdk2 activity on cell-cycle progression in mESCs. The shorter time-point (6 h) of olomoucine II treatment was chosen in order to follow the immediate effects of Cdk2 activity attenuation on mESC cycle progression. After the 6-h treatment, the cells were double-stained with BrdU and propidium iodide and analyzed by flow cytometry. The olomoucine II treatment increased the frequency of mESCs in G1 phase from 16% in mock-treated V6.5 cells up to 42% in 5 µM olomoucine II-treated V6.5 cells (P = 0.01) and from 26% up to 44% in HM1 cells (P = 0.04) (Fig. 4A).
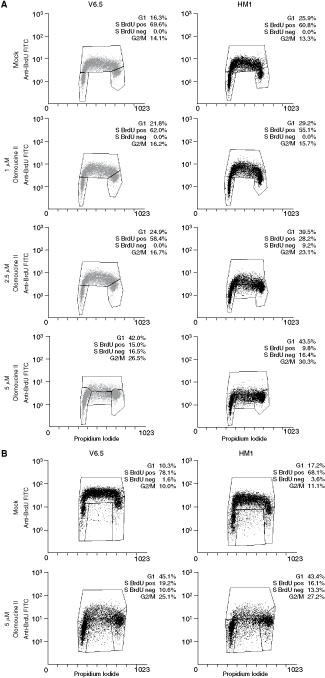
High Cdk2 activity is required for embryonic stem cell (ESC)-specific cell-cycle profile. Flow cytometry analysis of cell-cycle profiles and DNA replication in V6.5 and HM1 mouse (ESCs) treated with indicated doses of olomoucine II for 6 h (
Olomoucine II treatment also led to a decrease in S phase cell numbers and an inhibition of DNA replication. The S phase cell numbers decreased significantly in both V6.5 and HM1 cells treated with 5 µM olomoucine II (P = 0.03 and 0.04, respectively; Fig. 4A). Of the S phase cells, about 50%–60% did not incorporate BrdU, that is they were S phase arrested in both mESC lines after 5 µM olomoucine II treatment (P = 0.03 for V6.5 and 0.01 for HM1 cells; Fig. 4A). Lower doses of olomoucine II had no effect on DNA replication in V6.5 cells. DNA replication of HM1 cells appeared to be more sensitive to olomoucine II, as inhibition of DNA replication occurred (in 32% of S phase cells; P = 0.04) even under 2.5 µM olomoucine II treatment (Fig. 4A).
Prolongation of olomoucine II treatment to 11 h led to cell-cycle changes comparable to those after 6-h treatment (Fig. 4B): G1 phase cell numbers increased (P = 0.001 and 0.01 for V6.5 and HM1 cells, respectively), S phase cell numbers decreased (P = 0.002 for both V6.5 and HM1 cells), and G2/M phase cell numbers increased (P = 0.003 for V6.5 and P = 0.001 for HM1 cells) (Fig. 4B). Also, 5 µM olomoucine II treatment inhibited DNA replication in 40%50% cells (P = 0.02 for V6.5 cells and P = 0.01 for HM1 cells), as judged from the diminished numbers of BrdU-positive S phase cells (P = 0.008 and 0.003 for V6.5 and HM1 cells, respectively) (Fig. 4B). Treatment of mESCs with olomoucine II for longer periods of time (up to 96 h) sustained increased G1 phase cell numbers; however, a massive cell death could be observed (data not shown).
Olomoucine II treatment leads to G1 phase prolongation in mESCs
Our data showed that inhibition of Cdk2 activity by olomoucine II in mESC culture increases the proportion of cells in G1 (Fig. 4). This could be the result of either prolongation of the G1 phase, or arrest in the G1- or G1/S phase. To distinguish between these possibilities and to investigate the effect of Cdk2 inhibition on G1/S phase progression in mESC in greater detail, mESCs were synchronized in the G2/M phase by nocodazole treatment and then released from the block to obtain the maximum number of G1 phase cells, which were then followed in their cell-cycle progression under olomoucine II or mock treatment. The maximum number of G1 phase cells (41%) was observed at 2 h after nocodazole release, so at this time-point, olomoucine II was added to a final concentration of 5 µM. Olomoucine II-treated and mock-treated cells were collected at different time-points and their cell-cycle profile was analyzed by flow cytometry (Fig. 5). Olomoucine II treatment slowed down G1 phase progression and S phase entry (Fig. 5) in mESCs. The G1 phase was prolonged by ∼2 h (ie, to about 5 h), as shown by the fact that identical proportions of cells in G1 and S phases were found 4 h after nocodazole release in mock-treated cells and 6 h after nocodazole release in olomoucine II-treated cells, respectively. Collectively, these data suggest that a decrease in Cdk2 activity prolongs G1 phase duration from about 3 h in mock-treated cells to ∼5 h in 5 µM olomoucine II-treated cells.

Down-regulation of Cdk2 activity in mouse embryonic stem cells (mESCs) by olomoucine II leads to prolongation of the G1 phase and delayed S phase entry. V6.5 mESCs were synchronized by nocodazole treatment and 2 h after nocodazole release, the cells were treated with 5 µM olomoucine II or mock-treated. Cells were collected at indicated time-points after nocodazole release and their cell-cycle profiles were analyzed by flow cytometry. Hyperdiploid cells (>2n) were excluded from assessment of cell-cycle distribution.
Down-regulation of Cdk2 activity establishes a somatic cell-like cell cycle and induces expression of differentiation markers in mESCs
It has been shown that down-regulation of Cdk2 activity in hESCs induces their differentiation [18]. To find out whether this might also be the case in mESCs, we investigated the effects of prolonged Cdk2 down-regulation by olomoucine II in mESCs. After 24 h of 5 µM olomoucine II treatment, mESCs still exhibited a somatic cell-like profile, that is, a cell-cycle profile with a high proportion of cells in G1 phase and a lower proportion of cells in S phase (Fig. 6A). Changes in the mESC cycle induced by down-regulation of Cdk2 activity were accompanied by changes in cell morphology, cell and colony shape, and adhesiveness as early as 11 h after 5 µM olomoucine II treatment (Supplementary Fig. 1A). Examination of pluripotency and differentiation markers by quantitative real-time RT-PCR showed a statistically significant increase in expression of differentiation markers and reduced expression of some pluripotency markers after 11 h of olomoucine II treatment (Supplementary Fig. 1B). This was further reinforced by prolonged olomoucine II treatment for 24 h (Fig. 6B). Specific down-regulation of Cdk2 activity in mESCs reduced expression of the pluripotency marker Dax1 and induced expression of trophoblast (Cdx2), mesoderm (Brachyury), extraembryonic entoderm (Gata4), primitive endoderm (Lamb1-1), primitive neuroectoderm (Nestin), and skeletal (Runx2) markers (Fig. 6B). These data are indicative of induction of mESC differentiation by down-regulation of Cdk2 activity.

Down-regulation of Cdk2 activity by olomoucine II establishes a somatic cell-like cell-cycle profile and induces differentiation of mouse embryonic stem cells (mESCs). (
siRNA knockdown of Cdk2 increases G1 phase cell number, decreases S phase cell number, and induces morphological changes in mESCs
To verify that the observed effects of olomoucine II on mESC cycle regulation were truly caused by down-regulation of Cdk2 activity, we adopted Cdk2 siRNA knockdown approach. Twenty-four hours after Cdk2 siRNA transfection, Cdk2 expression was down-regulated by 43%, as assessed by quantitative real-time RT-PCR (Fig. 7A), and Cdk2 protein level decreased to 55% (Fig. 7B). These changes in Cdk2 expression led to 28% decrease of Cdk2 activity (data not shown) and induced increase of G1 phase cell number from 20.7% in negative control siRNA-treated cells to 26.3% in Cdk2 siRNA-treated cells (P = 0.01) (Fig. 7C), which was similar to the effect of olomoucine II on mESC cycle structure. Moreover, siRNA-mediated knockdown of Cdk2 induced similar changes in mESC morphology, colony shape, and adhesiveness (Fig. 7D) as olomoucine II (Supplementary Fig. 1A). These results confirm that Cdk2 is the target molecule for olomoucine II, through which the observed cell-cycle changes and associated modulation of pluripotency are mediated.
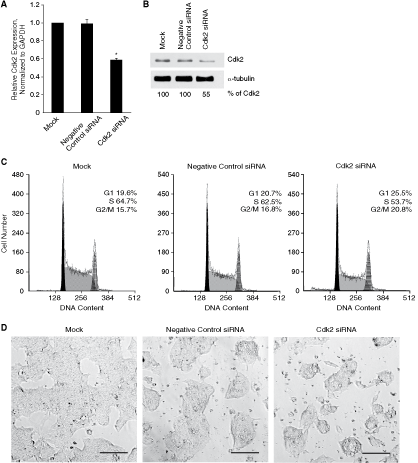
Cdk2 knockdown leads to similar mouse embryonic stem cell (mESC) cycle and morphology changes to those induced by olomoucine II treatment. (
Discussion
In mESCs, high, cell-cycle-independent Cdk2 activity was reported to underpin rapid cell-cycle progression [3] but the role of Cdk2 in the establishment of mESC-specific cell cycle has remained obscure. Moreover, in the context of Cdk substitutionary roles [15 –17] and Cdk2 nonessentiality for G1/S transition [13,14], the actual role of Cdk2 in mESC cycle regulation has not been fully elucidated. Finally, the role of Cdk2 in mESC cycle regulation was emphasized by the observation of a predominantly centrosomal localization of Cdk2 in mESCs (our observation, data not shown).
In our model, we used olomoucine II to down-regulate Cdk2 activity in mESCs. As olomoucine II is a Cdk inhibitor with high specificity for both Cdk2 and Cdk9 [20], we used also a Cdk9 inhibitor, CAN508 [21], to distinguish the effects of Cdk9 inhibition from those of Cdk2 inhibition. CAN508 is highly selective for Cdk9 and its effects on Cdk2 are minor. In in vitro kinase assays, CAN508 inhibits Cdk9 activity at least 50 times more effectively than it does Cdk2 activity [21].
mESC treatment with olomoucine II at a concentration equal to its IC50 caused major changes in cell-cycle progression: the G1 cell numbers increased and S phase cell numbers decreased (Fig. 1). CAN508 treatment caused similar, but smaller changes to those after olomoucine II treatment corresponding to its lower specificity for Cdk2. Olomoucine II treatment also led to a marked inhibition of DNA replication in mESCs. Overall, these cell-cycle changes in mESCs following olomoucine II or CAN508 treatment were clearly different from the effects of these inhibitors in somatic cells (Fig. 1).
Investigation of Cdk activities in inhibitor-treated mESCs confirmed Cdk9 inhibition in CAN508-treated cells and both Cdk2 and Cdk9 inhibition in olomoucine II-treated cells (Figs. 2 and 3). However, at higher concentrations of olomoucine II, Cdk1 inhibition occurs (Fig. 3) and might be partially responsible for the observed cell-cycle changes when 10 µM (or more) olomoucine is used. Therefore, we investigated cell-cycle progression of mESCs in 5 µM olomoucine II, that is in conditions of specific Cdk2 inhibition. Even under such specificity, G1 phase cell numbers in mESCs increased significantly with a concomitant reduction in S phase cell number and inhibition of DNA replication. In synchronized mESCs, we showed that the increase in the proportion of G1 phase cells after Cdk2 inhibition was caused by slowdown of their progression through G1 phase, that is prolongation of G1 phase by ∼2 h (from 3 to 5 h). This observation corroborates the role of Cdk2 in driving ESC cycles [3]. Of particular interest, our observation of distinctive cell-cycle changes after Cdk2 inhibition is novel and adds more depth to the observation of Stead et al. [3]. In their study, inhibition of Cdk2 activity (by treatment with a Cdk2 inhibitor Ro09-3033) did not change the general cell-cycle characteristics of mESCs, although they did observe slowdown of cell-cycle progression after Ro09-3033 treatment as we did after olomoucine II treatment. We suggest that high Cdk activity per se contributes to a very short G1 phase in mESCs and might be responsible for the unusual cell-cycle structure of mESCs.
As the atypical cell-cycle structure with short gap phases is common to all pluripotent cells of embryonic origin [3], it has been proposed that specific cell-cycle regulation and loss of G1 functions (restriction point and G1 checkpoint) in ESCs might be involved in self-renewal of ESCs [25]. The relationship between cell-cycle regulation and pluripotency of ESCs has been under dispute. It was reported that activation of p53 leads to rapid differentiation of ESCs by introducing changes in cell-cycle progression, particularly abolishing S phase entry [26]. Differentiation is a smart way of coping with DNA damage in ESCs [27], which are deficient in a G1 checkpoint response and p53-dependent apoptosis [28,29], by eliminating DNA-damaged ESCs from the replicative ESC pool. Recently, it was shown that down-regulation of Cdk2 activity in hESCs induces their differentiation to extraembryonic lineages [18]. Our study in mESCs presents further evidence on the requirement for Cdk2 activity for maintenance of the ESC-specific cell-cycle structure and for the pluripotency of ESCs. Down-regulation of Cdk2 activity in mESCs by olomoucine II or Cdk2 siRNA knockdown established a somatic cell-like cell cycle (Figs. 6A and 7C, respectively) and induced expression of differentiation markers of lineages of all 3 embryonic layers (Supplementary Fig. 1B and Fig. 6B). This observation is consistent with studies reporting cell-cycle remodeling during differentiation and an increase in G1 cell proportion with the loss of pluripotency [1,3,5,25].
Also, our study revealed slight differences in effects of Cdk2 down-regulation between mESCs and hESCs: while hESCs arrest in G1 after down-regulation of Cdk2 activity [18], mESCs only slowdown their G1 progression (Fig. 5). Neither after prolonged 5 µM olomoucine II treatment (48 or 72 h), nor after treatment with higher doses of olomoucine II (10 µM and higher) did we observe G1 arrest in mESCs (data not shown). Our contrasting observation could be caused by biological differences between mESCs and hESCs, but rather it might be related to the extent of Cdk2 inhibition or to different methods used for down-regulation of Cdk2 activity. In our study, we primarily analyzed requirements for Cdk2 in cell-cycle regulation of mESCs using a chemical inhibition approach. To confirm our observations, we also employed Cdk2 knockdown by siRNA. Both approaches led, in principle, to the same results, but in the knockdown experiments the G1 cell number was increased less efficiently. This might result from low efficiency of Cdk2 knockdown by siRNA; that is, from the different extent to which Cdk2 activity was inhibited. Another possibility is that the knockdown approach introduces a bias through up-regulation of other Cdks/cyclins and their potential compensation for S phase-promoting functions. Chemical inhibition might not allow for the same compensation [19]. While knockdown of a Cdk leaves a pool of its interactory cyclin molecules free and accessible for other Cdks that might bind to them and compensate for the knocked-down Cdk, chemical inhibition of a Cdk does not leave its partner cyclin pool available.
Nevertheless, whatever the modularities of induced cell-cycle changes in different ESC types are, down-regulation of Cdk2 activity commonly results in expression of differentiation markers. In this context, it would be interesting to investigate the effects of Cdk2 down-regulation in cancer stem cells. As cancer stem cells use an ESC-like “stemness” program to induce and maintain tumors [30,31], modulation of the cell cycle of (cycling) cancer stem cells with Cdk inhibitors might become a useful tool for their eradication.
Our study partially addressed the issue of testing Cdk inhibitors on an ESC model. Stem cells, in general, are more resistant to toxins and various types of drugs due to high expression of specific ATP-binding cassette (ABC) drug transporters [32]. As this characteristic might be common to “normal” (embryonic and tissue) stem cells and “abnormal” (cancer) stem cells [32], drug testing using ESCs as convenient model of cancer stem cells might provide important insights into the mechanism(s) by which cancer stem cells might response to cancer therapy.
We report on the cytotoxicity of olomoucine II and CAN508 in mESCs. Cytotoxicity of these drugs toward mESCs is similar to the cytotoxicity toward cancer cell lines, as the IC50 for olomoucine II or CAN508 was similar for mESCs and cancer cells. However, the mESC line V6.5 exhibited lower sensitivity toward CAN508, with an IC50 2 to 3 times higher than those for the mESC line HM1 (this study) and for cancer cell lines (reported previously [21]). We suggest this might be due to some unique features of transcription regulation in V6.5 cells [33], rather than due to ABC-mediated drug resistance, as V6.5 sensitivity toward another small molecular Cdk inhibitor, olomoucine II, does not significantly differ from that of other cell lines, including the mESC line HM1. We conclude mESCs do not exert resistance toward Cdk inhibitors olomoucine II and CAN508.
Besides other functions, cyclin-dependent kinases are critical regulators of cell-cycle progression and RNA transcription [34]. As a result of a variety of genetic and epigenetic events, Cdks are frequently deregulated in tumors [35,36]. Chemical Cdk inhibitors have appeared as a promising tool for cancer therapy because Cdk inhibition can lead to cell-cycle arrest and apoptosis [34]. But as Cdk2 activity is dispensable for cancer cell proliferation [37], the suitability of Cdk2 as a target for cancer therapy has been called into question. As modulation of Cdk2 and Cdk1 activity sensitizes survival checkpoint responses after exposure to DNA-damaging agents [38], Cdk2 inhibitors show promising antitumor activity in combination with DNA-damaging drugs [39]. Our study reveals olomoucine II as an effective tool for manipulation of ESC cycle and ESC pluripotency and in this context, it is tempting that Cdk2 inhibitors might become powerful tools for cancer stem cell eradication, possibly contributing to their differentiation.
Footnotes
Acknowledgments
The authors thank to Asst. Prof. Ladislav Dušek, Ph.D. (Institute of Biostatistics and Analyses, Masaryk University, Brno, Czech Republic) for statistical analysis. This work was supported by grants NR/9508 (Ministry of Health) and MSM 6198959205 (Ministry of Education, Youth and Sport). P.D. and V.K. were in part supported by MSM 6198959216 (Ministry of Education, Youth and Sport).