
Abstract
Umbilical cord blood (UCB), an ideal source for transplantable hematopoietic stem cells (HSC), is readily available and is rich in progenitor cells. Identification of conditions favoring UCB-HSC ex vivo expansion and of repopulating potential remains a major challenge in hematology. CD133+ cells constitute an earlier, less-differentiated HSC group with a potentially higher engraftment capacity. The presence of SCF, Flt3-L, and TPO are essential for CD133+ and/or CD34+ cells ex vivo expansion; however, IL-3 and IL-6 influence has not yet been clearly established. We investigated this influence on CD133+ cells from UCB ex vivo expansion and the effect of these cytokines upon cell phenotype. Immediately after isolation an 85% of CD133+ cell purity was obtained, diminishing after 4 and 8 days of ex vivo expansion. CD133+ fold-increase was higher using IMDM with SCF, Flt3-L, and TPO (BM)+IL-3 or BM+IL-3+IL-6 on day 8 (13.83- and 17.47-fold increase, respectively). BM+IL-6 presented no significant difference from BM alone. We demonstrated that 5.1% of the CD133+ cells expressed IL-6 receptor (IL-6R) after isolation. After 4 and 8 days in culture, the percentage of CD133+ cells that expressed IL-6R was as follows: BM alone (9.8% and 22.02%, respectively); BM+IL-3 (8.33% and 16.74%); BM+IL-6 (9.2% and 17.67%); and BM+IL-3+IL-6 (12.5% and 61.20%). Cell cycle analysis revealed quiescent cells after isolation, 95.5% CD133+ cells in the G0/G1 phase. Regardless of culture period or cytokine incubation, CD133+ cell cycle altered to 70% of CD133+ in the G0/G1 phase. Colony-forming unit (CFU) doubled in BM+IL-3+IL-6 after 8 days of incubation compared with BM group. SOX-2 and NANOG-relative gene expression was detected on day 0 after isolation. BM+IL-6 prevented the decrease in NANOG and SOX-2 gene expression level compared to BM+IL-3 or BM+IL-3+IL-6 incubated cells. Our results indicated that UCB-isolated CD133+ cells were better ex vivo expanded in the presence of SCF, Flt3-L, TPO, IL-3+IL-6. IL-3 probably promotes higher CD133+ cell expansion and IL-6 maintains immature phenotype.
Introduction
U
CD133+ cells are expressed in a subpopulation of CD34+ HSC derived from various sources including fetal liver and bone marrow, adult bone marrow, UCB, and mobilized peripheral blood [1 –4]. In addition to their hematopoietic capacity, these cells are capable of in vitro differentiation into neuronal cells [5], endothelial cells [6,7] and myoblasts [8], suggesting broader multipotential capacities of CD133+ populations compared to those of CD34+. However, the small number of CD133+ cells in a single cord blood unit limits their use in cell transplant procedures, thus ex vivo expansion may be useful to achieve a sufficient number of cells for clinical use.
Most ex vivo cultures of CD34+ and/or CD133+ cells from different sources use cytokine mixtures that include stem cell factor (SCF), thrombopoietin (TPO), and Flt3-L. For example, Ueda et al. showed that the addition of a complex composed of IL-6 and soluble IL-6 receptor (IL-6/sIL-6R) to SCF, TPO, and Flt3-L supports CD34+ cells expansion of human severe combined immunodeficient (SCID)-repopulating cells [9]. Gammaitoni et al. reported a striking increase of CD34+ cell number from cord blood cells after 16 weeks of culture [10]. Summers et al. reported a very high incidence of long-term culture-initiating cell (LTC-IC) and colony-forming cell (CFC) expansion in CD133+ cell cultures serum-free supplemented with SCF, TPO, and Flt3-L [11].
The role of IL-3 and IL-6 in CD133+ and/or CD34+ cells ex vivo expansion has not yet been clearly established. CD34+ cells from UCB expanded for 12 weeks, cultivated with SCF, TPO, and Flt3-L in the presence of IL-6, presented a more prominent LTC-IC expansion and NOD engraftment than cells cultured in the presence of IL-3 [12]. On the other hand, the presence of IL-3 instead of IL-6 showed an even better ex vivo expansion; the NOD/SCID engraftment however was less effective [13]. In addition, Encabo et al. demonstrated that the inclusion of IL-6 to SCF, TPO, and Flt3-L in a serum-free medium culture had a positive effect on the expansion of CD34+ and CD133+ cells from UCB, and in the number of primitive cells, detected by LTC-IC. However, the addition of IL-3 in the culture medium induced a negative effect on LTC-IC [14]. These results suggest a negative effect of IL-3 in the NOD/SCID engraftment and LTC-IC activity; this cytokine, however, presented a positive effect on cell expansion. On the other hand, the presence of IL-6 was not as efficient as IL-3 in the ex vivo expansion; this cytokine, however, increased the number of primitive cells, detected by LTC-IC. The mechanism of IL-3 and IL-6 in the expansion of ex vivo CD133+ cells and maintenance of primitiveness is not yet known. In addition, the phenotypic profile of CD133+ cells after expansion ex vivo has not yet been studied. Nevertheless, there are some data suggesting that CD133+ cells isolated from UCB express markers associated with both pluripotent and multipotent stem cells. Transcript analysis of freshly isolated CD133+ cells revealed the expression of pluripotency markers including Oct-4, Rex-1, LIFR, SOX-2 [15], and NANOG [16 –18].
Thus the aim of the present study was to investigate the influence of IL-3 and IL-6 or both on ex vivo expansion of CD133+ from UCB for 8 days and the effect of these cytokines upon cell phenotype analyzed by flow cytometry and real-time PCR.
Materials and Methods
Cell isolation
After informed consent from the mothers, UCB samples were collected from normal full-term deliveries, by standard procedures. Mononuclear cells were obtained by Ficoll-Hypaque (Amersham Pharmacia, Upsala, Sweden) density gradient centrifugation. CD133+ cells were isolated by immunomagnetic cell separation (MACS System; Miltenyi Biotec, Bergisch Gladbach, Germany), in accordance with the manufacture’s recommendations.
Recombinant cytokines
The following recombinant purified human cytokines used in this study were: stem cell factor (SCF), thrombopoietin (TPO), Flt3 ligand (Flt3-L), IL-3, and IL-6 (Petrotech Inc., New Jersey, USA), anti-CD133-PE (Ebioscience, San Diego, CA).
Expansion cell culture
After isolation, CD133+ cells were cultured in IMDM (Iscove’s modified Dulbecco’s medium) supplemented with 10% FBS, fungizone (2.5 μg/mL), penicillin (0.063 g/L), and streptomycin (0.1 g/L). Cells were seeded in 60-mm cell culture dish. Culture media were as followed:
Basal medium (BM): IMDM supplemented with 10% FBS, SCF (25 ng/mL), TPO (10 ng/mL), FLT3 (10 ng/mL)
BM+IL-3: BM + IL-3 (10 ng/mL)
BM+IL-6: BM + IL-6 (10 ng/mL)
BM+IL-3+IL-6: BM+IL-3 (10 ng/mL) + IL-6 (10 ng/mL).
Cultures were incubated at 37°C in 5% CO2 and 95% air in a fully humidified atmosphere for 8 days and medium was replaced every 4 days.
Flow cytometry
Flow cytometry analysis was performed on a FACSCalibur (Becton Dickinson, San Jose, CA) using the cell Quest software (Becton Dickingson). CD133+ cells were labeled with PE-anti-CD133 (Ebioscience) and FITC-anti-IL-6R α-chain1 (Santa Cruz) according to standard protocols. Apoptosis was assessed by flow cytometry using annexin-V binding. In brief, cultured CD133+ cells were washed in PBS and resuspended in annexin-V-binding buffer (BD Pharmingen, San Diego, CA, USA) 1 × 105 cells/mL; 100 µL of cell suspension was incubated with fluorescein isocyanate (FITC)-labeled recombinant annexin-V (BD Pharmingen) for 20 min, RT, in the dark. The samples were then diluted in binding buffer. Cell death was accessed by adding propidium iodide (PI) (0.025 μg) into annexin-V suspension.
Cell cycle analysis
CD133+ cells were cultured in different media and cell cycles were investigated. In brief, cells were fixed in ethanol 70%, incubated with a buffer, consisting of PI (10 μg/mL), Triton X-100 (0.01%), RNAase (0.1 mg/mL) for 20 min at room temperature. Flow cytometry analysis were performed on a FACSCalibur (Becton Dickinson) using the cell ModFit BDβ; Biotechnology software.
Colony-forming unit (CFU)
After 8 days of expansion in the different culture medium, total cells were adjusted and 1 × 104 cells were seeded in methylcellulose medium with recombinant cytokines without EPO (MethoCultTM Stem Cell Technology Inc., Vancouver, BC, Canada) for 14 days. All colonies were counted using an inverted light microscope. All values were normalized by the basal medium (BM) of each separate experiment.
Preparation of mRNA and real-time PCR: RNA extraction
Samples of CD133+ cells (up to 106 cells) after isolation from UCB or after 8 days of culture in specific culture medium were submitted to RNA extraction with RNAspin Mini RNA Isolation Kit from GE Healthcare™. The RNA samples were quantified using the Nano Drop Spectrophotometer (ND-1000®).
Reverse transcription
One microgram of each RNA sample was incubated with 1 U DNaseI (Life Technologies, Carisbad, CA) for 15 min at room temperature and EDTA was added to a final concentration of 2 mM to stop the reaction. The enzyme was subsequently inactivated for 10 min at 65°C. The DNaseI-treated RNA samples were reverse-transcribed with 200 U SuperScript III (Invitrogen) for 50 min at 50°C. Two units of RNaseH (Life Technologies) were subsequently added and the samples were incubated at 37°C for 20 min. The cDNA samples were quantified using the Nano Drop Spectrophotometer (ND-1000®).
Real-time PCR
Real-time detection of amplification was performed in a StepOnePlus TM Real-Time PCR (Applied Biosystems, Foster City, CA) using SybrGreen PCR Master Mix (Applied Biosystems). Approximately 15 ng of each cDNA sample were used in the reaction with the following primers: (1) 150 nM of ABL control forward and reverse primers (Fw: 5′-TGG AGA TAA CAC TCT AAG CAT AAC TAA A-3′; Rv: 5′-GAT GTA GTT GCT TGG GAC CCA-3′); (2) 300 nM of NANOG (Fw: 5′-CCAACATCCTGAACCTCAGC-3′; Rv: 5′-CCTTCTGCGTCACACCATT-3′); (3) 100 nM of SOX-2 forward and reverse primers (Fw: 5′-TAC CTC TTC CTC CCA CTC CA-3′; Rv: 5′-GGT AGT GCT GGG ACA TGT GA-3′). ABL expression was used as an endogenous control. The relative quantification value of NANOG and SOX-2 gene expression was calculated using the equation 2−ΔΔCT [19]. A negative control, “No Template Control,” was also included for each primer pair. The dissociation protocol was performed at the end of each run to check for nonspecific amplification. Two replicas were run on the same plate for each sample.
Results
IL-3 reduces the relative amount of CD133+ cells from UCB
After the isolation of CD133+ cells by immunomagnetic assay, flow cytometry was carried out and the purity observed was ∼85% (Fig. 1A).
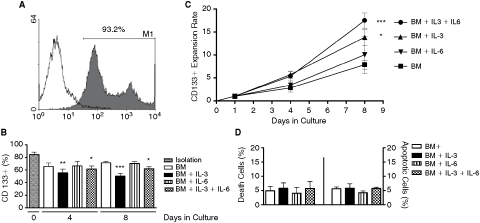
IL-3 and IL-6 influence on CD133+ cell expansion. (
Upon cultivation in a defined basic medium (BM) (supplemented with 10% SBF and growth factors such as SCF, Flt3-L, and TPO), the IL-3 or IL-6 or both cytokines were added in order to investigate the percentage and expansion rate of the CD133+ cells. Flow cytometry analyzed on days 4 and 8 revealed that the percentage of CD133+ cells diminished in the group of cells incubated with BM alone (65.55% and 71.39%); BM+IL-3 (56.64% and 50.50%); BM+IL-6 (66.53% and 70.22%); and BM+IL-3+IL-6 (61.67% and 61.95%), respectively, after 4 and 8 days (Fig. 1B). More interesting, the addition of only IL-6 to the medium did not statistically change the percentage of CD133+ cells (66.53% and 70.22%). Thus, an important observation is that after isolation, the percentage of CD133+ cells decreased ∼24% in all groups and during the entire time frame observed (Fig. 1B). The reduction was significant when IL-3 alone or IL-6 was added to BM (Fig. 1B). These results suggest that CD133+ cells lost CD133 expression during the culture and this loss was more prominent using IL-3.
IL-3 and IL-6 are necessary for better ex vivo expansion of CD133+ cells from UCB
In spite of the loss of CD133 expression, an expansion of these cells was observed. The absolute amount of CD133+ cells was estimated on day 0 after isolation, days 4 and 8 of culture, and the fold-increase was calculated as follows: CD133+ number on day 4 or 8 divided by CD133+ number on day 0 (Fig. 1C). The fold-increase of cells incubated with BM+IL-3 and BM+IL-3+IL-6 was statistically higher on day 8 (13.83- and 17.47-fold increase, respectively). On the other hand, CD133+ cells cultured with BM+IL-6 for 4 and 8 days did not present significant differences compared with BM (10.2- and 7.85-fold increase, on the 8th day, respectively). To discharge the possibility that the low fold-increase of cells incubated with BM+IL-6 was due to cell death or apoptosis, we proceeded with annexin/PI flow cytometry. As demonstrated by Figure 1D, a basal percentage of cell death and apoptosis (<10%) was observed in all groups studied.
As previous studies demonstrated that CD133+ cells isolated from cord blood cells expressed IL-3 receptor [20], further experiments were carried out to determine the expression status of IL-6 receptor (IL-6R) on CD133+ cells and characterize which population of CD133+ cells was actually expanded. Flow cytometry analysis demonstrated that 5.1% of the CD133+ cells expressed IL-6R after isolation. After 4 and 8 days in culture, the percentage of CD133+ cells that express IL-6R was as follows: BM alone (9.8% and 22.02%); BM+IL-3 (8.33% and 16.74%); BM+IL-6 (9.2% and 17.67%); and BM+IL-3+IL-6 (12.5% and 61.20%), respectively (Fig. 2).

Expression status of IL-6R during CD133+ cell ex vivo expansion. Representative dot plot of CD133+ and IL-6R expression after isolation from Umbilical cord blood (UCB), 4 and 8 days in 4 different culture media as indicated. Numbers correspond to percentages of CD133+ cells (upper right quadrant), CD133+IL-6R+ (upper left quadrant), and CD133negIL-6R (down right quadrant). Abbreviation: BM, basal medium.
Clonogenic activity of CD133+ cells improved in the presence of IL-3 and IL-6
Colony-forming unit (CFU) assay was used to identify the clonogenic capacity of CD133+ cells after ex vivo expansion in the current culture conditions. We counted the total CFU number, indistinguishable from CFU types (CFU-mix). We demonstrated that cells incubated with BM+IL-3+IL-6 for 8 days doubled the numbers of CFU-mix (statistically significant) when compared with cells cultured in BM (Fig. 3). Although IL-3 and IL-6 did not induce a statistically significant increase in the number of CFU, we observed a tendency toward augmentation.
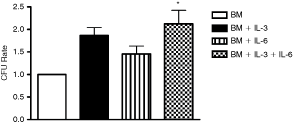
Colony-forming units assay after CD133+ cell ex vivo expansion. After expansion time (8 days) with 4 different culture media, 1 × 104 cells were seeded in methylcellulose and the numbers of global CFU subtypes were analyzed after 14 days. All values were normalized by the basal medium (BM) of each separate experiment. Values are the mean ± SEM (n = 3). Each experiment was performed in duplicate. *P < 0.05 when compared to BM group (one-way ANOVA test followed by Bonferroni’s test).
CD133+ cell cycle status is not influenced by IL-3 and/or IL-6
Cell cycle analysis revealed that after isolation, 95.5% of CD133+ cells were in the G0/G1 phase, 1.33% S phase, and 3.27% G2-M, characteristically as quiescent cells (Fig. 4A). Regardless the period of culture (4 or 8 days ex vivo expansion) or cytokine incubation (IL-3, IL-6, or both), ∼70% of CD133+ cells remained in the G0/G1 phase, whereas 30% CD133+ cells were in the S phase and 3% in the G2-M phase (Fig. 4B–4E). Thus, these results suggest that IL-3 and/or IL-6 did not interfere in the cell cycle status when compared to the BM at these time points. In addition, the transference of these cells from cord blood to culture condition induced a change in the percentage of G0 phase to S phase of ∼30%, regardless of the cytokine used, suggesting a deviation of cells from G0/G1 phase to S phase (Table 1).

CD133+ cell cycle status analyzed in different culture medium CD133+ DNA content. CD133+ cell cycle status analyzed in different culture medium CD133+ DNA content. (
C
CD133+ cell cycle analysis after 4 days in culture. CD133+ cell cycle status was investigated immediately after CD133+ isolation and in the different groups after 4 and 8 days in culture. Data were analyzed by flow cytometry. Data are given as percentage mean ± SEM of 4 different experiments.
Abbreviations: BM, basal medium; IL, interleukin.
IL-6 maintains NANOG and SOX-2 gene expression of CD133+ cells during ex vivo expansion
SOX-2 and NANOG are not only essential for pluripotency and self-renewal of embryonic stem cells but also expressed in somatic stem cells that have superior expansion and differentiation potentials [21 –24]. The relative gene expression of SOX-2 and NANOG was quantified by real-time PCR as shown in Figure 4. We observed a higher relative gene expression of SOX-2 and NANOG on day 0 after isolation. Indeed, BM+IL-6 prevented the decrease in gene expression levels of NANOG and SOX-2, when compared to cells incubated with BM+IL-3 or BM+IL-3+IL-6 (Fig. 5). In addition, ex vivo expansion with BM, BM+IL-3, and BM+IL-3+IL-6 showed higher decrease in relative gene expression levels of SOX-2 and NANOG, when compared with gene expression observed on day 0 after isolation.
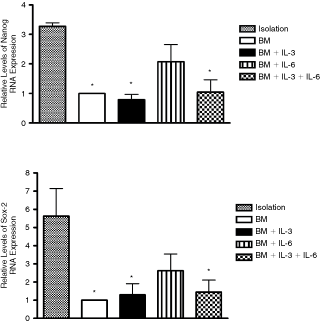
Real time after CD133+ cell isolation and ex vivo expansion. RNA extractions were carried out immediately after isolation of CD133+ cells and after 8 days of CD133+ cells ex vivo expansion. mRNA was later on transcripted to cDNA for quantitation by real-time PCR. Relative levels of mRNA expression were compared against the data of CD133+ cells after isolation. Values are expressed as mean ± SEM (n = 3). All data were normalized by the basal medium (BM) of each separate experiment. *P < 0.05 (one-way ANOVA test).
Discussion
The mechanisms responsible for ex vivo expansion of CD133+ cells have not yet been adequately elucidated. In the present study, CD133+ cell culture for 8 days in basic medium containing IL-3+IL-6 showed a more prominent cell proliferation and clonogenic activity, compared with cells cultured in basic medium exclusively, basic medium+IL-3 or basic medium+IL-6. On the other hand, IL-6 alone prevented a decrease in the gene expression of SOX-2 and NANOG, at the same level as gene expression of CD133+ cells right after isolation. Neither IL-3 nor IL-6 interfered in the cell cycle status.
Many investigators have used various combinations of cytokines that act upon primitive HSC in order to optimize ex vivo expansion culture conditions. The combination of cytokines ex vivo is used, as the development of hematopoietic stem/progenitors cells (HSPC) is believed to be regulated, at least in part, by interactions of cytokine receptor signals. In particular, SCF and Flk2/Flt3 ligand have been used as key cytokines for HSPC expansion, as c-Kit and Flk2/Flt3-L, tyrosine kinase receptors for SCF and Flt3-L, respectively, were shown to transduce crucial signals for HSPC development [25 –29]. TPO, a ligand for c-Mpl, originally identified as a primary regulator for megakaryopoiesis, has also been shown to stimulate the expansion of primitive hematopoietic cells [30 –32].
Differently from SCF, Flt3-L, and TPO, the utilization of IL-3 in the ex vivo expansion of HSPC has been considered as an attractive possibility; however, the mechanism of action remains complex and controversial. Binding of IL-3 to its receptor molecules results in their dimerization that activates a ras-mediated (resraf-1-MAP kinase) pathway [33,34] and a ras-independent JAK (janus kinase)-STAT (signal transducer and activator of transcription) system [35,36]. The action of IL-3 has been studied using either cell lines or hematopoietic cells recovered from bone marrow, peripheral blood, or cord blood. IL-3 enhances the survival of HSC and stimulates their proliferation and differentiation [37] and also favors, under certain conditions, their self-renewal [38]. Our present data demonstrated that IL-3 or IL-3+IL-6 decreased the relative amount of CD133+ cells after 4 and 8 days of expansion; however, IL-3 or IL-3+IL-6 induced a higher expansion rate after 8 days in culture, as shown by the increased absolute number of CD133+ cells. Cell death or apoptosis were not induced by IL-3, IL-6, or with both together. Our results corroborate the data obtained by Encabo et al. These data suggest that, though able to induce CD133+ expansion, IL-3 may induce loss of the phenotype of some CD133+ cells [14]. The intracellular signaling pathway that involves these effects has not yet been elucidated; however, Bonati et al. observed that massive proliferation and self-renewal capacity of CD34+ cells isolated from UCB after addition of IL-3 was independent from sustained ERK phosphorylation [39]. Data from literature demonstrated that in early and late anaphase, CD133 protein, also called prominin-1, was enriched toward the cleavage furrow, and in some cases, also remained clustered at one pole of the diving cells, probably causing an equally or unequally distribution between the 2 nascent daughter cells [40,41]. Considering these results, we hypothesized that the presence of IL-3 could induce an asymmetric distribution of CD133 molecules between daughter cells, as IL-3 and IL-3+IL-6 induced a decrease in the CD133+ percentage, as demonstrated in our study.
Concerning IL-6, the effects observed were the opposite. The relative amount of CD133+ cells was higher than in the presence of IL-3, suggesting that IL-6 may prevent loss of CD133+ phenotype. IL-6 binds to the IL-6 receptor and the complex of IL-6 and IL-6R associates with the signal transducing membrane protein gp130 promoting dimerization that leads to the activation of the intracytoplasmic JAK tyrosine kinases [42]. The signaling–transducing via gp130-JAK, activated by IL-6, plays an important role in the stem cell biology [43]. Our data demonstrated that IL-6R is expressed in the membrane of CD133+ cells isolated from UCB and that after 4 and 8 days of expansion in all medium conditions, the most pronounced expression was observed in the CD133+ cells incubated with BM+IL-3+IL-6 for 8 days. Our present data corroborated the data obtained by Campard et al. who demonstrated that the expression and production of IL-6R by CD133+ isolated from peripheral blood were induced by incubation with SCF, Flt3-L, EPO, IL-3, IL-6, and/or hyper-interleukin-6 (a complex of IL-6 and soluble IL-6R). The increased expression of mIL-6R led to a better response of IL-6 in the survival and proliferation of CD133+ cells [44]. Thus, the phenotype maintenance of CD133+ cells by IL-6 may be mediated by interaction of IL-6 to its receptor expressed by these cells.
An important observation is that the percentage of CD133+ cells slightly decreased after 4 or 8 days in culture, independently from the presence of IL-3 or IL-6 in the culture. On the other hand, the expansion rate of CD133+ cells in the presence of IL-6 was lower than the expansion rate observed in CD133+ cells incubated with IL-3. Confirming these data, Encabo group also demonstrated that IL-6 slightly influenced the CD133+ percentage or cell expansion rate. Concerning the effect of IL-6 upon transplantable human cord blood cells expansion, Bruno et al. demonstrated that IL-6 has no effect on long-term maintenance or even expansion of CD34+ cells isolated from cord blood cells and cultured over 6 weeks [13]. Apparently, IL-6 alone did not influence the percentage and ex vivo expansion of CD133+ cells according to the present data. On the other hand, recently published data demonstrated that a serum-free culture of the CD133-sorted human cord blood cells in the presence of the combined use of FP6 (artificially generated IL-6/sIL-6R fusion protein that efficiently transmit gp130 signals in HSC) together with SCF, TPO, and Flt3-L expanded the number of cells in 6-fold, probably due to gp130-mediated signals playing a critical role in human transplantable HSC [9,45]. The lack of IL-6 effect in the expansion rate of CD133+ observed in this study may be attributed to an insufficient gp130 signaling, and further studies should be carried out.
In this study, the clonogenic activity of CD133+ cells ex vivo expanded was maximum on day 8 of cell culture with basic medium+IL-3+IL-6. The same result was observed by Suzuki et al. who also demonstrated that the replacement of IL-6 by FP6 (artificially generated IL-6/sIL-6R fusion protein) in the basic medium containing IL-3 generated an even higher number of CFU [45]. Taking these results together, we can suppose that IL-6 improves clonogenic activity of CD133+ cells ex vivo expanded.
As shown herein, CD133+ cells freshly isolated from UCB shifted their cell cycle phase from quiescent cells to cells that show a high DNA synthesis, indicating proliferative cells (during the period of ex vivo expansion). Similar data was demonstrated by Grskovic et al. [46]. In addition, we demonstrated that cell cycle status of CD133+ cells was not influenced by the addition of IL-3, IL-6, or both together for 4 and 8 days of ex vivo expansion. Recent studies demonstrated that stem cell activity resides in the CD133+ G0 population, and this population has a highly repopulating activity when compared with their G1 counterparts [47] and a greater capacity of CFU formation [11].
Recent data demonstrated that NANOG might be a key gene required to maintain the pluripotency state in murine embryonic stem cells [48,49]. In addition, Boyer et al. revealed a close relationship between Oct-4, NANOG, and SOX-2 evaluated in human embryonic stem cells, which appeared to interact with feedback regulatory circuits [50]. Concerning gene expression in CD133+ cells from UCB, the results obtained by Baal et al. suggested that these cells might have a greater potential of differentiation as they expressed different pluripotent markers including SOX-1, SOX-2, FGF-4, Rex-1, and Oct-4 [18]. Finally, we performed real-time PCR to investigate the phenotype of CD133+ cells after 8 days of ex vivo expansion in the presence or absence of IL-3 or IL-6 and we demonstrated that BM+IL-6 prevented the decrease in gene expression levels of NANOG and SOX-2. In addition, Encabo et al. demonstrated that IL-6 prevented the differentiation induced by IL-3 on expansion of CD34+ cells from UCB [14]. These results together suggest that IL-6 may preclude the down-regulation of SOX-2 and NANOG, maintaining CD133+ cells in a pluripotent state when expanded ex vivo in the presence of IL-6.
In conclusion, CD133+ cells isolated from UCB are better expanded ex vivo in the presence of SCF, Flt3-L, TPO, IL-6+IL-3, as IL-3 probably promotes a higher expansion and IL-6 maintains the phenotype.
Footnotes
Acknowledgments
We are grateful to Fernanda Gonçalves Pereira Cunha for the assistance in FACS analysis. We would also like to thank the group of the Cord Blood Bank for providing the UCB samples. Tereza Sueko Ide Salles for her serious effort in the Laboratory. Financial support: FAPESP and CNPq.