
Abstract
Coordinated transcription factor networks have emerged as the master regulatory mechanisms of stem cell pluripotency and differentiation. Many stem cell-specific transcription factors, including the pluripotency transcription factors, OCT4, NANOG, and SOX2 function in combinatorial complexes to regulate the expression of loci, which are involved in embryonic stem (ES) cell pluripotency and cellular differentiation. This review will address how these pathways form a reciprocal regulatory circuit whereby the equilibrium between stem cell self-renewal, proliferation, and differentiation is in perpetual balance. We will discuss how distinct epigenetic repressive pathways involving polycomb complexes, DNA methylation, and microRNAs cooperate to reduce transcriptional noise and to prevent stochastic and aberrant induction of differentiation. We will provide a brief overview of how these networks cooperate to modulate differentiation along hematopoietic and neuronal lineages. Finally, we will describe how aberrant functioning of components of the stem cell regulatory network may contribute to malignant transformation of adult stem cells and the establishment of a “cancer stem cell” phenotype and thereby underlie multiple types of human malignancies.
Introduction
T
Pluripotent stem cells possess the unique ability to self-renew and differentiate into all of the cell lineages present in the embryo and adult. ES cells are pluripotent cells derived from the inner cell mass of the early stage blastocyst (please refer to reference [1] for a thorough review of the history of stem cell research and the molecular characteristics of undifferentiated stem cells) (Fig. 1A). The vitamin A metabolite, all-trans-retinoic acid can induce the differentiation of both mouse and human ES cells [2,3]. Mouse [4,5] and human [6] ES cells share this extraordinary differentiation capacity and possess similar, though not identical, molecular characteristics [7]. In the absence of serum, mouse ES cells require both the leukemic inhibitory factor (LIF) and bone morphogenetic proteins (BMPs) for the maintenance of the undifferentiated state [8]. These features of mouse ES cell culture requirements were central to elucidating the role of STAT3 [9], BMP-SMAD [8,10], and the inhibitor of differentiation (ID) [11] pathways in the maintenance of mouse ES cells. In the presence of serum, activation of STAT3 by LIF-cytokine is sufficient to maintain mouse ES cells in an undifferentiated state [9,12]. However, this is not the case for human ES cells [13]. These differences in tissue culture requirements likely contributed to the challenges in derivation of pluripotent human ES cells, which were overcome in 1998 by Thomson and colleagues, who reported the successful isolation and characterization of human ES cells [6]. In contrast to mouse ES cells, BMPs induce differentiation [14] and STAT3 activation by LIF is not sufficient to maintain self-renewal of human ES cells [13]. Suppression of BMP signaling by basic fibroblast growth factor (bFGF) contributes, but is not sufficient, to maintain human ES cell self-renewal [14,15]. Interestingly, mouse pluripotent stem cells derived from the E5.5-E6.5 postimplantation embryos (epiblast stem cells, Epi-SCs) fail to self-renew in the presence of LIF or BMP4; however, they share the requirement of FGF4 and activin/nodal signaling for self-renewal [16,17], as is the case for human ES cells. This suggests that self-renewal and pluripotency in human ES cells may be a more complicated process that requires more cooperative factors compared with that in mouse ES cells. Stem cell lines derived from the mouse blastocyst, in the presence of FGF, activin, and the Wnt pathway activator, BIO [7], termed FAB-SCs, share characteristics of both Epi-SCs and human ES cells and express the pluripotency factors Nanog, Oct4, and Sox2, but are not pluripotent [18]. Yet, treatment of FAB-SCs with LIF/BMP4 induces latent pluripotent capacity in these cells, which is attributable to the induction of E-cadherin expression [18]. These results indicate that parallel molecular pathways exist in mouse and human ES cells through which cells can achieve similar transcriptional and epigenetic states consistent with pluripotency. Indeed, it now appears that in the absence of any extrinsic signal, self-renewal is the default program of ES cells [19].
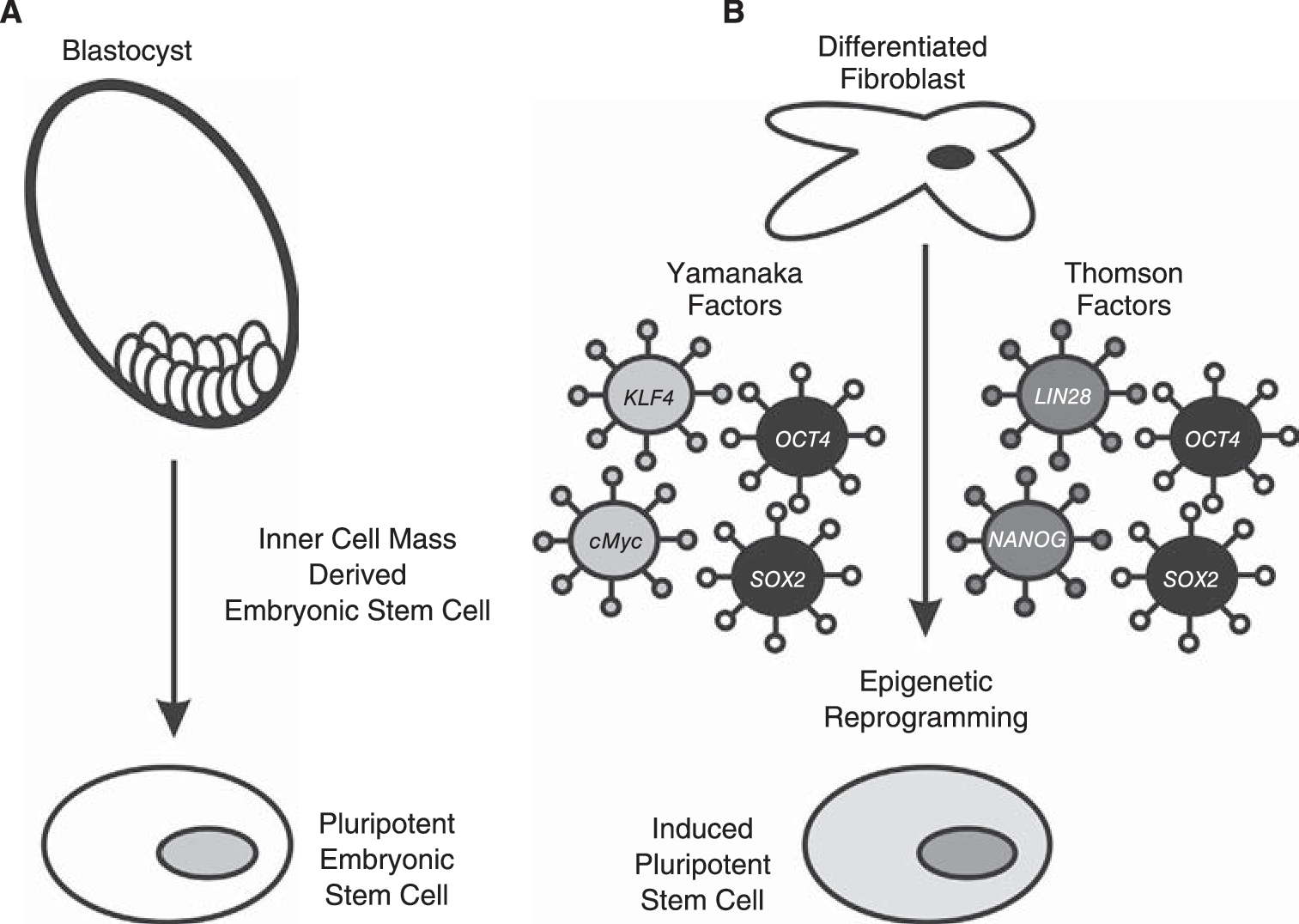
Pluripotent stem cells can be derived from cells isolated from the inner cell mass of early stage blastocysts (
Further insight into the critical signaling determinants of pluripotency were revealed by the experimental induction of pluripotency by somatic cell nuclear transfer [20,21], nuclear reprogramming/cell fusion experiments [22 –24], and most recently by retroviral introduction of 4 critical genes, now sometimes referred to as the Yamanaka factors: Oct4, Sox2, Klf4, and c-Myc (Fig. 1B), or a combination of OCT4, SOX2, NANOG, and LIN28 [25]. This technique has permitted the reprogramming of multiple distinct mouse and human differentiated cell types to yield iPS cells [26 –34]. The highest efficiencies of induced pluripotency are achieved when all 4 factors were utilized; however, c-Myc [35] and Klf4 [36] have been shown to be dispensable for somatic cell reprogramming to pluripotency under specific culture conditions. Specifically, the histone deacetylase inhibitor valproic acid (VPA) both enhances the efficiency of iPS derivation by the combined 4 factors and permits the derivation of iPS cells using just Oct4 and Sox2 [37]. These studies indicate that Oct4 and Sox2 are critical factors required for maintaining self-renewal and pluripotency of mouse and human stem cells. Indeed Oct4 was sufficient to induce pluripotency in adult neural stem cells, which express endogenous Sox2, c-Myc, and Klf4 [38], whereas Klf4 could induce pluripotency from Epi-SC [39]. In the context of this review, these experiments have been pivotal in revealing the importance of OCT4 in pluripotency and that each pluripotency transcription factor possesses a unique epigenetic function to influence pluripotency and differentiation of stem cells. Recent advances in nonviral methodologies to introduce reprogramming factors into differentiated cells represent major advances toward the ultimate clinical application of these iPS cells [40 –42]. A related but distinct combination of 4 critical genes, OCT4, SOX2, NANOG, and LIN28, can also induce pluripotency of human somatic cell types [25]. OCT4 and SOX2 are central to the iPS methodologies described by the Yamanaka-laboratory [26,27,29 –31,35]. Later in this review we will address the pivotal functions of the NANOG homeodomain protein in pluripotency [43,44] and the recently described roles of LIN28 in regulation of microRNA processing in stem cells [45]. Further recent studies have determined that the timing of expression of pluripotency-associated factors appears to directly influence the generation of iPS cells. A combination of OCT4, NANOG, SOX2, LIN28, c-MYC, and KLF4 enhances iPS derivation by a factor of ∼10, and reduces the time of reprogramming from 26 to 17 days [46]. Recently, Zhao and colleagues reported that a timely knockdown of p53 (p53-siRNA) combined with forced expression of UTF1 was able to increase the efficacy of iPS formation by ∼100-fold, in a background consisting of fibroblasts pretransduced with the classic Yamanaka factors [47]. These remarkable studies reinforce the concept of pluripotency as a reprogrammable state established as the outcome of a transcriptional circuit involving key stem cell transcription factors and microRNAs.
Epigenetic events involving reversible histone modifications and DNA methylation govern stem cell self-renewal and differentiation
The crucial role of epigenetics in modulating the transcriptional outcome and thereby regulating cell fate decisions has emerged over the last decade. Epigenetics can be defined as “heritable” or transmitted changes in the chromatin structure independent of the underlying DNA sequence. These changes include a functionally diverse array of distinct covalent histone modifications [48] and methylation of the DNA CpG islands [49] (Fig. 2). Although DNA methylation primarily mediates transcriptional repression [49], different histone modifications such as acetylation, methylation, phosphorylation, and ubiquitination play a more complex role in regulating gene transcription [50]. The transcriptional status of a locus/gene, whether it is expressed or repressed, is modulated by local histone covalent modifications. Indeed the specific histone residues modified and the nature of the covalent modification constitute a histone code, which segregates the genome into regions which are active, repressed, or are poised for activation [48]. Several studies have provided compelling evidence that distinct chromatin organization and epigenetic signatures exist within ES cells and govern their intrinsic ability to self-renew and differentiate into multiple lineages [51 –61]. An elegant study reported by Meshorer and colleagues [62] revealed hyper-dynamic association of architectural chromatin proteins (including heterochromatin protein-1 and histones) within ES cell chromatin, as compared to lineage committed neural progenitor cells (NPCs) [62]. Additionally, overall chromatin architecture, as assessed by heterochromatin organization, differed in ES cells as compared to the NPCs. While the heterochromatin pattern was more dispersed in ES cells, it formed compact and discrete foci in NPCs [62]. There have also been reports of global increases in acetylation of histone 3 (H3) and histone 4 (H4) in ES cells indicative of an open chromatin structure [63]. However, histone acetylation is dynamic during stem cell differentiation and this reflects the important roles of histone deacetylases (HDACs) in guiding stem cell differentiation and embryonic development [64,65]. Histone deacetylase I (HDAC1), the mammalian homolog of yeast RPD3, functions to deacetylate core histones and thereby contributes to chromatin condensation and transcriptional repression. HDAC1 is also implicated as a key regulator of genome activation in 1-cell embryos [65]. HDAC1 also participates in the nucleosome remodeling–histone deacetylase (NuRD) complex that plays essential roles in stem cell biology [66]. HDAC1, and the closely related HDAC2, are also the enzymatic components of the distinct Nanog–Oct4–deacetylase (NODE) complex, which functions to control developmentally regulated and pro-differentiation genes [64]. Consistent with this critical function, an HDAC1−/− null mutation in mice results in embryonic lethality because of severely retarded proliferation and development [67,68]. HDAC1 is also found in complexes with the YinYang1 (YY1) polycomb transcription factor and other polycomb complex members, including the EED/EZH2 components of the polycomb repressive complex-2 [69]. HDAC1 can also interact with the de novo DNA methyltransferase 3a (Dnmt3a) [70,71] and with Ddx5/p68 [67], establishing a link between epigenetic transcriptional regulation and the RNA helicases, which play diverse roles in miRNA biogenesis and transcriptional regulation [72] (see section on microRNAs). Thus, HDAC1 contributes to distinct and dynamic transcriptional repressive complexes with essential roles in stem cell differentiation and development.

Bivalent chromatin domains, composed of activating (histone H3K4 trimethylation) and repressive (H3K27 trimethylation) histone tail modifications (indicated in gray) are a hallmark of developmentally regulated genes. Transcriptional repression of pro-differentiation genes is maintained in pluripotent stem cells by polycomb repressive complexes (PRC) 1 and 2. The core PCR2 includes the EZH2 H3K27 methyltransferase, SUZ12, and EED. However, HDAC1 and YY1 have been shown to interact with PRC2. PRC1 includes Bmi1, the Ring1A/B ubiquitin ligases, and mammalian homologs of Drosophila polyhomeotic (Ph) and polycomb (Pc). Transcriptional repression can be relieved by the combined actions of the JMJD3 and UTX H3K27 demethylases. DNA CpG methylation also contributes to the developmental regulation of gene expression. Individual CpG islands may be unmethylated (○), partially/hemi-methylated (), or fully methylated (•).
Recent advances in the use of combined genome microarray and chromatin immunoprecipitation (ChIP-chip) technology have enabled researchers to study patterns of genome-wide histone modifications. This technology has proven essential for the identification of the characteristic “epigenetic signature” of pluripotent stem cells. Bernstein and colleagues used a combination of ChIP and oligonucleotide tiling arrays spanning ∼2.5% of the mouse genome comprising of a subset of highly conserved noncoding elements to identify the histone modification patterns in mouse ES cells [59]. This study reported a novel chromatin pattern, “bivalent domain” that harbors both the “repressive” trimethylated histone H3 lysine 27 (H3K27me3) and “activating” trimethylated histone H3 lysine4 (H3K4me3) modifications [59] (Fig. 2). The majority of these bivalent domains were enriched at the promoters of genes encoding transcription factors that regulate developmentally important genes in pluripotent stem cells. A subsequent study utilized ChIP and high throughput DNA sequencing techniques (ChIP-Seq) to compare the chromatin states (including methylation of H3K4, H3K27, H3K36) of mouse ES cells, neural progenitors, and embryonic fibroblasts [73]. Comparison of the genomic distribution of these histone modifications and their correlation with the CpG content of 17,762 mouse promoters in undifferentiated and differentiated cell types studied permitted the identification and segregation of promoters as active, repressed, or poised for distinct developmental fates. Bivalent chromatin domains are also present in human ES cells [54,55]. The large scale of these studies enabled the identification of distinct categories of genes that exclusively possess the H3K4me3 or H3K27me3 marks, genes containing a combination of H3K4me3 and H3K27me3, and genes that lack both modifications (see Table 1 for examples). The activating H3K4me3 modification is associated with genes implicated in essential biological functions such as cell proliferation and metabolism. Consistent with the reports on the mouse epigenome, combinations of H3K27me3 and H3K4me3 were found to be associated with genes involved in important developmental functions such as neurogenesis, ectoderm formation, and transcriptional regulation [54,55]. Notably, the Hox cluster family was present in the class of genes with colocalized H3K4me3 and H3K27me3 epigenetic marks in both mouse and human studies. The Hox family of transcription factors has an essential role in axial patterning during embryogenesis, and Hox genes are well-established targets of polycomb group (PcG) proteins [58,74,75]. Similarly, promoter regions of developmentally important genes occupied by LIF-activated STAT3 in mouse ES cells exhibit these bivalent chromatin states [76]. This suggests that integration of additional signaling pathways is necessary to convert extrinsic LIF signaling into meaningful epigenetic and developmental decisions in mouse ES cells. Therefore, there is convincing evidence that bivalent modification in ES cells is associated with repression of genes involved in development, possibly due to the PcG-mediated repressive H3K27me3 mark. In addition, it is likely that these genes are “poised” for activation, most likely due to the presence of the H3K4me3 mark. However, how these opposing modifications are incorporated at these bivalent sites in ES cells remains to be determined. Another key question pertains to the precise function of these bivalent domains. It is still unclear if the existence of such domains is solely important for proper differentiation or if they play a role in establishment and/or maintenance of pluripotency. Although initial studies suggested that bivalent domains were possibly a feature unique to stem cells, several differentiated cell lineages have been shown to possess bivalent sites. These include mouse neuronal precursor cells, mouse embryonic fibroblasts, human T cells, and human lung fibroblasts [77 –79]. Together, these studies have identified the existence of a dynamic chromatin architecture in undifferentiated ES cells, which undergoes global reorganization during differentiation.
To illustrate the convergence of pluripotency transcription factor networks with epigenetic processes, we selected a subset of ES cell genes identified as Oct4, Nanog, and Sox2 target genes in both human (Boyer et al., 2005) and mouse ES cells (Kim et al., 2008)
This dataset was then examined in independent datasets for promoter occupancy by Oct4, Nanog [99] and for Nanog, Oct4, SOX2, TCF4 and polycomb complexes [144], bivalent histone methylation marks (K4 and/or K27), and DNA promoter methylation classification [87]. *Zfp42/Rex1 is included for comparison (see Fig. 3).
Abbreviations: TF, transcription factor; N, Nanog; O, OCT4/Pou5f1; S, SOX2; T, TCF; –, not detected; Suz12/EZH2, PRC2; Ring1b, PRC1; K4, histone H3 lysine 4 trimethylation; K27, histone H3 lysine 27 trimethylation; HCP, high CpG promoter containing a 500 bp region with GC content ≥0.55, observed to expected CpG ratio ≥0.6; LCP, low CpG promoter containing no 500 bp region and with observed to expected CpG ratio ≥0.4 and ICP, intermediate CpG promoter, CpG distribution between HCP and LCP; ESC, embryonic stem cell; NPC, neural precursor cell.
DNA methylation and stem cell differentiation
DNA methylation (Fig. 2) is an epigenetic modification typically associated with gene silencing (reviewed in detail in [49]). Methylation of DNA CpG islands plays a critical role in physiological processes of transcriptional repression, X-inactivation, and genomic imprinting [80]. Three different DNA methyltransferase (Dnmt1, Dnmt3a, Dnmt3b) isoforms have been characterized. Dnmt1 exhibits a substrate preference for hemimethylated CpG and functions primarily to maintain DNA CpG methylation, whereas Dnmt3a and Dnmt3b are critical for de novo DNA methylation [80,81] (Fig. 2). Dnmts play essential functions in embryonic differentiation and development [52,82,83]. However, the exact roles of DNA CpG methylation in stem cell differentiation and how this may relate to other epigenetic regulatory mechanisms remain poorly understood. The genome-wide DNA methylation profiles analyses have revealed that DNA CpG methylation is a dynamic epigenetic trait resulting in distinct patterns of methylated CpG regions in undifferentiated stem cells and cells undergoing differentiation [52]. Interestingly, DNA methylation is associated with the majority (87%) of ES cell-repressed genes, which lack the bivalent histone domain [84]. This finding suggests that DNA methylation is a potential repressive mechanism for this class of genes, which is in a silenced state in ES cells (see Table 1). DNA methylation can also contribute to the silencing of the key pluripotency transcription factors as ES cells move toward a differentiated state. Dnmt3a and Dnmt3b were shown to methylate Oct3/4 and Nanog promoters in embryonal carcinoma and ES cells upon differentiation [81]. Consistent with this, the expression of several genes associated with pluripotency, including Nanog 1 and Zfp42/Rex1, is characterized by demethylated promoters in ES cells and the genes are silenced and methylated in differentiated mouse fibroblasts [84]. This reflects the distinct global DNA methylation patterns that contribute to stem cell differentiation. Nonetheless, the functional interplay between DNA methylation and histone modifications remains poorly understood. Indeed, the correlation between DNA methylation and PcG-mediated repression has been debated. Comparison of the genes targeted by PcG complex and those enriched by methyl-DNA immunoprecipitation revealed that polycomb-mediated repressive marks and DNA methylation are not associated with a common pool of target genes in ES cells [84]. Although there have been reports of functional interdependence of these epigenetic mechanisms [84,85], these and other results [86,87] suggest that DNA methylation and PcG-mediated transcriptional repression represent 2 distinct but convergent epigenetic silencing mechanisms.
The NANOG/OCT4/SOX2 pluripotency transcription factor network control embryonic stem cell differentiation by epigenetically modulating the expression of pro-differentiation loci
The transcription factors Oct4, Nanog, and Sox2 play essential roles in the regulation of pluripotency in both human and mouse ES cells [43,88 –90]. Down-regulation of the Oct4 protein leads to a loss of maintenance of ES cell pluripotency as the cells differentiate [43,89]. Surprisingly, however, the overexpression of Oct4 also induces differentiation in ES cells [88]. This suggests that precise levels of Oct4 must be sustained for the maintenance of pluripotency, further implying that Oct4 levels are tightly regulated by the cell. Nanog is a homeodomain protein that is predominantly expressed in pluripotent cells and is essential for early embryo development [43]. Evidence from the derivation of iPS cells (described earlier) and other studies [43,89] indicates that while Nanog is not required for the establishment of pluripotency in ES cells, it does function to maintain the self-renewal capacity of these cells and its expression has been shown to suppress differentiation [31,44,91]. Consistent with this role, Nanog overexpression enables the propagation of mouse ES cells in a leukemia inhibitory factor (LIF)-free environment [43] and human ES cells in the absence of feeder cells [92]. Conversely, the loss or deficiency of Nanog results in ES cell differentiation [93,94]. Nanog [64], like Oct4 [95], forms distinct transcriptional repressive complexes, but more recent work has shown that Nanog possess 2 potent transcriptional activating domains [96,97]. This suggests that like Oct4 [98], Nanog can function as a transcriptional activator. The Sox2 transcription factor is a member of the SRY-related HMG box (Sox) transcription factor family [90,97]. Sox2 is expressed in pluripotent cells and multipotent embryonic and extraembryonic cells. Although Sox2 remains less well characterized than either Oct4 or Nanog [43,88,89], Sox2 is known to play a major role in the regulation of cell fate and with Oct4 appears essential for the derivation of iPS cells [36].
Oct4 and Nanog have long been recognized to be master transcriptional organizers of ES cell self-renewal [53,74, 96,99 –101]. It is clear that Oct4 and Nanog form a mutual, interdependent transcriptional network with Sox2. Oct4 and Sox2 have been known to act synergistically to regulate their own transcription [102] and as well as the expression of other key stem cell genes including FGF4 [103], Nanog [104], and Zfp42/Rex1 (Fig. 3) [98,105,106]. The pluripotency transcription factors can form combinatorial complexes that include Nanog homodimers [107] and heteromeric complexes including OCT4–SOX2 [104], Oct4–Nanog [108], and Nanog–Sall4 [109] proteins. As described earlier, the levels of Oct4 must be precisely maintained [88]. The exact mechanisms regulating OCT4 levels in ES cells remain to be determined, but appear to involve direct regulation by Sox2 [90]. Furthermore, Oct4 has been shown to suppress its own expression [100]. This Oct4 self-suppression creates a negative feedback loop to counter the actions of Sox2 and Nanog, thereby fine-tuning the levels of Oct4 present in the undifferentiated stem cells [100]. One functional consequence of fluctuating Oct4 levels is seen on the expression of Nanog. Oct4 actively regulates Nanog in a biphasic manner such that lower Oct4 levels up-regulate Nanog, whereas higher Oct4 levels result in down-regulation of Nanog expression [99].
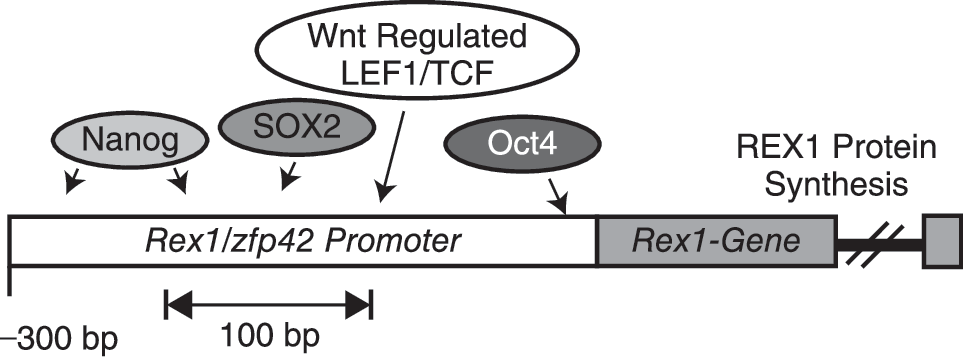
Convergent stem cell regulatory networks control expression of key stem cell genes. One classical representative of this transcriptional network is the Zfp42/Rex1 gene. Expression of the Zfp42/Rex1 gene has long been used as a marker of undifferentiated stem cells and is regulated by Nanog, Sox2, and Oct4, and by the Wnt pathway. Expression of Zfp42/Rex1 is also subject to epigenetic regulation by polycomb complexes and DNA methylation (see Table 1).
The pluripotency transcription factors function collaboratively to regulate the state of differentiation of ES cells. Two major studies have demonstrated that Oct4, Nanog, and Sox2 share a substantial fraction of target genes and, in fact, co-occupy genes in both mouse and human ES cells [74,99]. As reported by Boyer and colleagues, these genes occupy collectively about 10% of the promoters in the human genome. About half of the promoter regions bound by OCT4 were also bound by SOX2 and 90% of these doubly bound genes were in turn bound by NANOG [74]. Moreover, the OCT4, SOX2, and NANOG-binding sites were in close proximity, further confirming that the proteins work in concert. Data from mouse ES cells also indicate that Nanog and Oct4 colocalize in many gene regions [99] (see Table 1). Also, stem cell-associated genes were activated by Oct4 and Nanog, while inactive genes typically were implicated in cell lineage determination and development. Both studies also found that Oct4 and Nanog bound to gene regions associated with loci encoding microRNAs. However, in no case was a common microRNA identified as a potential Nanog or Oct4 target [74,99]. This may reflect intrinsic differences in the gene expression profiles of mouse and human ES cells or may be attributable in part to the different experimental techniques, ChIP-chip and ChIP-PET (paired-end ditag sequences), employed in the 2 studies.
The use of biotinylated-tag approaches has permitted biochemical characterization of the Nanog complex [101] and analysis of stem cell promoter occupancy of 9 stem cell transcription factors (including Oct4, Nanog, Sox2, c-Myc, Klf4, Zfp42/Rex1) [53]. These studies demonstrated that Nanog, its target genes, and protein partners interacted with a large number of proteins involved in early development in vivo. This method demonstrated that Oct4, Nanog, Sox2, and other associated proteins simultaneously occupy the promoters of a substantial number of genes. This supports the hypothesis that ES cell-associated transcription factors function as protein complexes. Genes whose promoters were bound by several transcription factors tended to be activated in ES cells, including the stem cell markers, Nanog, and Rex1. In contrast, genes involved in differentiation and therefore repressed in ES cells have promoters that tended to be bound by just one factor. Moreover, the authors indicated that this pluripotency protein network is also associated with other complexes such as PRC1, which functions in the repression of transcription [109 –111]. In conclusion, Oct4, Nanog, Sox2, and a number of associated transcription factor proteins such as Sall4 [109,110] activate and maintain the expression of genes involved in self-renewal, while simultaneously repressing genes that mediate differentiation. Thus Oct4, Nanog, and Sox2 form a self-reinforcing and intricately connected network that preserves ES cell character. Hence, elucidating the various interactions between these proteins and their targets and protein partners remains a critical step in enhancing our understanding of the molecular basis of pluripotency.
Polycomb group proteins
Polycomb group (PcG) proteins play essential roles in epigenetic regulation of ES cell differentiation and development [56,57,75,112]. PcG proteins can cooperate with OCT4 and NANOG in the regulation of differentiation [58,75]. Important members of the polycomb family of proteins include the enhancer of zeste-1 and 2 (EZH1 and 2) histone H3K27 methyltransferases [113], embryonic ectoderm development (EED), suppressor of Zeste (SUZ12), B lymphoma Mo-MLV insertion region 1 (BMI1), and Ring1A/B. The zinc finger protein YinYang1 (YY1) can recruit polycomb complexes to chromatin [69] and on this basis is considered a polycomb protein. PcG proteins form multimeric PRC [75,114] (Fig. 2). The exact subunit and isoform composition of each PRC can vary dependent upon cellular conditions and differentiation [115 –118]. In general, the EZH2 containing PRC2, which also requires SUZ12 and EED, is the complex that initiates polycomb repression. Distinct PRC2-related complexes with variant EED isoforms and/or SirT1 deacetylases have also been reported and exhibit distinct histone substrate specificities [115]. Polycomb epigenetic repression is maintained by PRC1, a large multi-subunit complex containing chromodomain proteins (CBC2/4/8), BMI1, and Ring1A/B ubiquitin-ligase activity (reviewed in [119]). YY1 can interact with the EED [120] and EZH2 [69] components of PRC2. YY1 can also interact with PRC1 under certain conditions [121].
The PRC2 proteins, EZH2, EED, and SUZ12, occupy and repress the promoter/regulatory domains of genes that participate in differentiation and cell fate decisions [57]. As described earlier, PcG proteins are also implicated in the establishment of “bivalent chromatin domains” that possess both repressive (histone H3 lysine 27 trimethylation; H3K27me3) and active (histone H3 lysine 4 trimethylation; H3K4me3) histone marks. Genes located within these bivalent chromatin domains, though epigenetically repressed, still possess the capacity for transcriptional activation in response to a differentiation stimulus [54,59,73]. During retinoic acid-induced differentiation of mouse teratocarcinoma cells, retinoic acid acting via its receptors causes PRC2 complexes to exit gene promoters transcriptionally activated by RA [122,123]. Thus PcG proteins have the epigenetic authority to maintain repression of developmentally regulated genes and thereby contribute to cell fate decisions [124].
The gene clusters repressed by the PcG complex change during differentiation. As repression of pro-differentiation loci is relieved, PcG-mediated epigenetic constraints are imposed on pluripotency genes such as OCT4 [54 –56,112,125]. The mechanisms controlling this reciprocal regulatory network and the sequence of events that balance pluripotency versus differentiation remain unknown. Expression of the Suz12 [56,112], Yy1 [126], and Ezh2 [127,128] polycomb genes is necessary for embryonic development. However, a recent study shows that Eed, and by extension its PRC2 partners, Ezh2 and Suz12, are not required to maintain ES cell pluripotency, but are necessary to repress expression of lineage-specific genes [129]. Thus PRCs can be considered to regulate patterns of differentiation (see Table 1).
Histone lysine methylation can be reversed by an enzymatically diverse group of transcriptional coregulators. JMJD3 and UTX are H3K27me3-demethylases that reverse PcG-mediated repression of Hox genes in mammalian ES cells upon differentiation [130,131] (Fig. 2). Given the ability of JMJD3 to reverse the repressive epigenetic mark (H3K27me3), which is associated with repression of several differentiation genes in ES cells, it would be reasonable to speculate about a role for Jmjd3 in activation of pro-differentiation genes. Indeed Jmjd3 is essential for commitment of ES cells to the neural fate [132]. Jmjd3 mediates the transition to the neural fate by activating key neurogenesis genes namely, Nestin, Pax6, and Sox1. The increased expression of neural programming genes correlates with the loss of the H3K27me3 repressive mark via the catalytic activity of Jmjd3 [132]. JMJD3 has also been shown to regulate the activation of epidermal differentiation genes in primary human keratinocytes, thus expanding their role in regulating differentiation in epidermis, yet another self-renewing tissue [133].
The Jmjd1a and Jmjd2c histone lysine H3K9me3-demethylases also play important roles in mediating the epigenetic regulation of pluripotency in mouse ES cells and are directly regulated by Oct4 [134]. Depletion of Jmjd1a and Jmjd2c by siRNA in mouse ES cells led to morphological changes, induced cellular differentiation, and increased expression of different lineage-specific markers [134]. Subsequent microarray analysis in combination with functional assays revealed downstream targets of these demethylases. Jmjd1a was found to be associated with the promoter of the Tcl1 gene, which encodes the Tcl1 cofactor of the Akt1 kinase. Tcl1 regulates ES cell self-renewal [135]. Furthermore, Jmjd2c associates with the Nanog promoter and thereby regulates the expression of Nanog [134].
MicroRNAs and stem cells
Recent studies have revealed that microRNAs (miRNAs), a family of nonprotein encoding transcripts of ∼20–25 nucleotides, play essential roles in regulating gene expression (see [136,137] for recent expansive reviews of miRNA biogenesis and function). A subset of miRNAs is preferentially expressed in undifferentiated stem cells [138] and plays essential roles in proliferation, pluripotency, and differentiation [139,140]. The pattern of miRNA expression changes during ES cell differentiation [141]. Mouse ES cells deficient in the Dicer [142] and Microprocessor (Drosha-DGCR8-Ddx5) [72,143] components of the miRNA processing apparatus exhibit defects in ES cell differentiation and development. The promoter regions of miRNAs that function in stem cells are typically occupied by key members of the stem cell transcription factor network, including Nanog, Oct4, and Sox2 [144,145]. Interestingly, Nanog, Oct4, and Sox2 are themselves subject to miRNA [146] and PcG [112] modulation during ES cell differentiation. A recent study showed that RA-induced differentiation of mES was associated with an up-regulation of miRNAs (miR-134, miR-296, miR-470), which target the coding sequences of the Nanog, Oct4, and Sox2 pluripotency transcription factors [147]. The miR-124 contributes to neuronal differentiation [148]. Collectively, these reports highlight the emerging view that miRNAs regulate, and are themselves regulated by, distinct transcriptional repressive mechanisms [45,144]. The best characterized miRNA network involves the ES-specific miR-290 cluster, which regulates Oct4-methylation in differentiating ES cells [149]. This illustrates that miRNAs play an indirect role in the control of de novo DNA CpG methylation in differentiating ES cells [149].
Convergence of distinct epigenetic repressive pathways and stem cell differentiation
There is evidence of functional convergence between the epigenetic repressive mechanisms, mediated by the PRC, DNA methyltransferase-3 (Dnmt3)-mediated promoter methylation, miRNAs and Oct4/Nanog, which participate in regulating ES cell differentiation [52,150,151]. ES cell chromatin is poised to initiate broad ranging and extensive transcriptional responses to pro-differentiation cues [59,125]. Therefore, one common effect of each of these parallel transcriptional repressive mechanisms is to reduce transcriptional “noise” [152]. This dampening down of “background” transcriptional events that could adversely bias patterns and timing of differentiation is achieved by a complex inter-regulation of the pluripotency network [153,154]. For example, expression of the EZH2 component of the polycomb repressive complex-2 is directly regulated by the miRNA-101 [155]. Conversely, the SUZ12 component of the polycomb complex and associated histone H3K27 methylation epigenetic mark were found to be associated with a subset of miRNAs inactive in undifferentiated ES cells [144]. The expression of these miRNA loci increases as the polycomb-mediated repression is relieved enabling these miRNAs to guide ES cell differentiation [144]. The YY1-PcG transcription factor also directly regulates transcription of the miR-29 miRNA by recruiting HDAC1 and EZH2 to regulatory regions in the miR-29 promoter region [156]. Similarly, miRNAs appear to regulate the stable silencing of pluripotency-associated genes, such as Oct4, by de novo DNA methylation [149]. Oct4 in turn regulates expression of the Eed component of the PRC2 [157] and recruits PRC1 to target loci silenced in undifferentiated stem cells [151,158]. Thus the NANOG, OCT4, SOX2 stem cell transcription factors, miRNAs, polycomb, and DNA methyltransferase complexes cooperate in a complex mutual regulatory circuit during ES cell differentiation (Figs. 2 and 4). The recently identified convergence of Oct4–Sox2 and miRNA pathways in the regulation of the ES cell cycle [159] suggests that the complexity of these networks is only now being deciphered.
As noted earlier, the Zfp42/Rex1 gene is a convergence point of critical stem cell differentiation pathways. Zfp42/Rex1 is regulated by Nanog [105], Sox2 [105], and Oct4 [98,99,106,160] (Fig. 3). Expression of Rex1 is also influenced by the Wnt pathway [7]. REX1 is conserved in human and mouse stem cells [161]. We [161] and others [162] have shown that the ZFP42/REX1 stem cell protein is a member of the YinYang1 (YY1) subfamily of zinc finger transcription factors, which includes the Drosophila polycomb protein, pleiohomeotic (PHO). The exact mechanism of how PRCs are targeted to the correct DNA regions remains poorly understood. Polycomb response elements (PREs) have been identified in the Drosophila genome [163]. However, no such features have been definitely identified in the mouse or human genomes. Pho is involved in the recruitment of polycomb complexes to response elements in Drosophila [163 –165]. YY1 has been shown to compensate partially for loss of Pho function in Drosophila [166]. This indicates that YY1, and by extension REX1, could play an analogous role targeting PcG complexes to specific loci in higher organisms. Studies have revealed that expression of Rex1 is limited to human [101,167 –171] and mouse [171] ES cells, a variety of adult stem cells [172,173] and iPS cells derived from distinct mouse and human somatic cell types [26,28,31,35]. Following stem cell differentiation, the Rex1/Zfp42 locus is subject to extensive histone H3 lysine 9 dimethylation, a G9a-mediated epigenetic mark consistent with tissue-specific transcriptional repression [51] and DNA methylation [84]. Biotin-affinity labeling of the REX1 protein has revealed that the REX1 interactome involves potential interactions with other key stem cell proteins, including OCT4 [101]. The functional significance of these potential REX1 interactions remains unexplored. More recently, the genome-wide distribution of 9 stem cell transcription factors, including Rex1, was analyzed by bioChIP-on-Chip in mouse ES cells [53]. This study revealed that Rex1 promoter occupancy most frequently clustered with epigenetic marks consistent with transcriptional activation (histone H3 lysine 4 trimethylation; H3K4me3). These studies have confirmed the importance of Rex1 in epigenetic regulation in undifferentiated stem cells. The effects of ectopic expression of REX1 in ES cells have also been studied [174,175]. Zhang and colleagues found that Rex1 reduced the self-renewal capacity of mouse ES cells, suggesting that Rex1 plays a role in regulating the self-renewal potential of cells in which it is expressed, whereas Masui and colleagues saw little effect [174,175]. These reports may reflect differences in the levels of REX1 expressed in the distinct experimental systems used in the studies [174,175]. These results may indicate that the ability of stem cells to self-renew is sensitive to subtle differences in Rex1 protein levels, as is also the case for Oct4 [88]. We have found that in the presence of retinoic acid, Rex1 null ES cells display an increased tendency to differentiate along all 3 germ lines, suggesting a role for Rex1 in retinoic acid-induced differentiation [176].

The reciprocal regulatory circuit composed of Oct4–Sox2–Nanog; polycomb repressive complexes and microRNAs regulate the transcriptional responses necessary to balance self-renewal and differentiation.
Transcriptional networks and lineage determination during stem cell differentiation
The functional interplay of each component of the stem cell regulatory circuit (Fig. 4) is perhaps best understood in the establishment of neuronal and hematopoietic lineages. Mouse embryonic stem cells can be effectively guided into becoming hematopoietic progenitors in serum-free media when exposed to BMP4, activin A, bFGF, and VEGF consecutively [177]. When co-cultured with murine stromal cells human ES cells can undergo definitive hematopoiesis [178], generating an easily accessible population for the dissection of transcription factors involved in lineage commitment. The accessibility of human blood has also enabled the identification of miR-155 as key regulators in the erythroid and myeloid lineage differentiation responses [179]. Similarly, in vitro neuronal differentiation protocols have been reported (refer to [77] for an example of human neural progenitor derivation from ES cells in a defined system). Mohn and colleagues have characterized the epigenetic changes that occur upon differentiation of pluripotent ES cells toward neuronal lineages [77]. This study revealed that the genome-wide distributions of H3K27me3 (a surrogate mark of polycomb repression) and DNA CpG methylation exhibited stage-specific distributions during differentiation from ES cells to neural progenitors to terminally differentiated pyramidal neurons [77]. Another key epigenetic regulator of neuronal differentiation of stem cells is RE1-silencing transcription factor (REST) [180,181]. Although a potential central role for REST in the maintenance of stem cell self-renewal and pluripotency remains controversial [182 –184], experimental depletion of REST by siRNA in ES cells impairs neuronal differentiation and development [185]. REST coordinates the epigenetic changes necessary for neural differentiation by interacting with and targeting HDAC1/2 and histone lysine demethylase 1 (LSD1/AOF2) to appropriate loci [186], during lineage commitment and neuronal differentiation [180,181]. Expression of REST is itself subject to miRNA regulation by miR-9/miR-9*, which are regulated by REST [187]. Thus, REST regulates neuronal differentiation by forming a reciprocal regulatory circuit composed of miR-NAs and diverse epigenetic mechanisms.
Stem cell transcriptional networks and human cancer
Small numbers of stem cells are believed to exist in most if not all adult tissues (reviewed in detail in [188]). Adult stem cells can evade the stringent genetic controls of their normal pathways of cellular differentiation and proliferation and can give rise to cancer. Cancer stem/initiating cells have been defined as a subset of cancer cells that have the exclusive ability of self-renewal and cause the heterogeneous lineages of cancer cells that comprise the tumor [189,190]. These “cancer stem cells” are implicated in cancer initiation, malignant potential, metastatic progression, and in the posttreatment recurrence of many human cancers types [191]. Stem cell-specific proteins, including OCT4, NANOG, and ZFP42/REX1 are implicated in some cancers [161,192,193]. Histologically poorly differentiated tumors showed preferential overexpression of genes normally enriched in ES cells. Activation targets of NANOG, OCT4, SOX2, and c-MYC are more frequently overexpressed in poorly differentiated tumors than in well-differentiated tumors [194]. It appears that the genes active in both ES cells and cancer stem cells are controlled by a few master regulatory genes, one of which is c-MYC. As noted earlier, c-MYC can contribute to epigenetic reprogramming and induction of pluripotency from differentiated fibroblasts (see section on iPS cells). However, c-MYC is also sufficient to reactivate the ES cell-like program in cancer cells [195]. These results suggest that aberrant activation of an ES cell-like transcriptional program in adult differentiated cells may induce pathological self-renewal characteristic of cancer stem cells.
There is also conclusive evidence implicating aberrant polycomb function in malignancy [196]. As described earlier, polycomb complexes contribute to the epigenetic regulation of key gene networks involved in stem cell self-renewal [197], differentiation, and proliferation and achieve this via dynamic regulation of chromatin/histone modifications (eg, via lysine methylation) associated with the promoter and regulatory regions of polycomb target genes [56,57,112,114,198,199]. This polycomb epigenetic stem cell gene signature has also been observed in cancer cells [200] and PRCs play important roles in cancer stem cells [201]. There is a wealth of evidence that overexpression of the EZH2 polycomb gene occurs in multiple human malignancies (eg, see [196,202]). Indeed genomic loss of miR-101 leads to increased EZH2 levels [155,203]. Although the exact mechanisms by which EZH2 contributes to carcinogenesis remain poorly defined, recent evidence indicates EZH2 overexpression can contribute to the inappropriate silencing of tumor suppressor genes [114]. For example, the pro-differentiation tumor suppressor gene, retinoic acid receptor β2 (RARβ2) was shown to be a PcG target [57], providing a plausible link between polycomb and epigenetic regulation of this important tumor suppressor gene whose expression has been found to be reduced or lost in many in human malignancies. For these reasons understanding the mechanisms by which the complex interplay of the pluripotency transcription factors, PRC, and miRNAs balance self-renewal and cellular proliferation (Fig. 4) is essential for our understanding of both the earliest events of embryonic differentiation and human carcinogenesis.
Footnotes
Acknowledgments
This research was supported by DOD-PCRP WXWH-081-0317 (V.K.), HHMI Gilliam Fellowship (N.C.R.); NIH-NRSA F31CA123703-02 (K.B.S.), NIH-NRSA T32MH018882 (S.M.S.), NIH R01CA097543, and NIH R01CA043786 (L.J.G.). This investigation was supported by grant UL1RR024996 of the Clinical and Translation Science Center at Weill Cornell Medical College (Pilot Award, N.P.M.).
Author Disclosure Statement
No competing financial interests exist.