
Abstract
Recent evidence shows that amniotic fluid (AF) contains multiple cell types derived from the developing fetus, and may represent a novel source of stem cells for cell therapy. In this study, we examined the paracrine factors released by human amniotic fluid–derived mesenchymal stem cells (AF-MSCs) and their ability to accelerate the wound-healing process by stimulating proliferation and migration of dermal fibroblasts. AF-MSCs expressed the typical MSC marker proteins CD13, CD29, and CD44 and differentiated into adipocytes, osteoblasts, and chondrocytes when exposed to the appropriate differentiation media. In addition, AF-MSC-conditioned media (AF-MSC-CM) significantly enhanced proliferation of dermal fibroblasts. Antibody-based protein array and enzyme-linked immunosorbent assay (ELISA) indicated that AF-MSC-CM contains various cytokines and chemokines that are known to be important in normal wound healing, including IL-8, IL-6, TGF-β, TNFRI, VEGF, and EGF. Application of AF-MSC-CM significantly enhanced wound healing by dermal fibroblasts via the TGF-β/SMAD2 pathway. Levels of p-SMAD2 were increased by AF-MSC-CM, and both the increase in p-SMAD2 and migration of dermal fibroblasts were blocked by inhibiting the TGF-β/SMAD2 pathway. Moreover, in a mouse excisional wound model, AF-MSC-CM accelerated wound healing. These data provide the first evidence of the potential for AF-MSC-CM in the treatment of skin wounds.
Introduction
M
Amniocentesis is a widely accepted procedure used in prenatal testing, and presents a low risk for both the mother and the fetus. Surplus AF from the procedure can be used to isolate AF-derived stem cells, which may represent a viable source of cells for therapeutic applications and thus avoid the ethical problems associated with other sources of fetal stem cells. AF is known to contain multiple cell types derived from embryonic tissues during the process of fetal development and growth. Cells that express Oct4, a marker of undifferentiated stem cells, were identified in human AF, and it has been proposed that AF might be an ideal source of pluripotent stem cells [2]. Subsequent studies developed a 2-stage culture protocol to isolate AF-derived MSCs (AF-MSCs) from second-trimester amniocentesis samples without interfering with the process of fetal karyotyping [3]. AF-MSCs have been further characterized and evaluated for their potential for differentiation into diverse cell types, including those of adipose, muscle, bone, endothelial, and neuronal lineages [4,5].
Recent studies in models of myocardial infarction, acute kidney failure, and stroke have shown that MSC therapy has the potential to inhibit cell death and stimulate endogenous regeneration programs [6 –8]. The effect of MSC transplantation is thought to be mediated by an increase in cell protective, angiogenic, and mitogenic factors, in addition to the differentiation of transplanted MSCs into specific cell types [9,10]. In fact, it has also been shown that MSCs secrete a number of cytokines and growth factors, including hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), and insulin-like growth factor-1 (IGF-1) [7,10 –12]. Moreover, it was recently demonstrated that systemic administration of MSC-conditioned medium (MSC-CM) significantly improved angiogenesis, cardiac protection, and short-term survival in a rat model of fulminant hepatic failure, and attenuated hypoxic pulmonary vasoconstriction [9,13,14]. In addition, MSC-CM inhibited apoptosis induced by hypoxia and serum starvation in vitro, as indicated by a decreased number of TUNEL-positive cells and caspase-3 activity and an increase in activation of the survival factor, AKT [15]. Furthermore, MSC-CMs promoted wound healing by stimulating the migration of dermal fibroblasts following skin injury [16]. These results suggest that in addition to direct participation in tissue reconstruction, MSCs are able to interact with and influence the activity of other cells.
Dermal wound healing is a very orderly and efficient process characterized by 4 distinct but overlapping phases: hemostasis, inflammation, proliferation, and remodeling [17]. The wound-healing response begins the moment the tissue is injured. In the early stages of wound healing, fibroblasts interact with surrounding cells that initiate numerous cell-signaling cascades, resulting in the synthesis of extracellular matrix (ECM), glycoproteins, adhesive molecules, and various growth factors. During the proliferative phase of wound healing, fibroblasts infiltrate the wound area and begin to deposit new ECM. The newly deposited collagen matrix then becomes cross-linked and organized during the final remodeling phase [18]. Thus, extensive proliferation and migration of fibroblasts is closely linked with granulation-tissue formation, the first manifestation of the early matrix, and is also associated with deposition of collagen fibers at later stages of the wound-healing process [19]. Possible approaches to enhance this process include the use of various growth factors and their combinations as possible therapies for wound healing in experimental and clinical studies [20 –23]. Several growth factors and cytokines, including platelet-derived growth factor (PDGF), transforming growth factor-β (TGFβ), keratinocyte growth factor (KGF), and insulin have been demonstrated to have beneficial effects on the wound-healing process [24].
The goal of this study was to characterize isolated AF-MSCs and their effect on a cutaneous wound-healing model. For this purpose, we isolated AF-MSCs and analyzed their capacity to differentiate into multiple cell types in vitro. Moreover, the secretion profiles of AF-MSCs were examined by analysis of AF-MSC-CM with cytokine array and enzyme-linked immunosorbent assay (ELISA). Finally, we investigated the effect of AF-MSC-CM on dermal fibroblast proliferation and migration in vitro, as well as its effect on wound healing following skin injury in vivo.
Materials and Methods
Culture and establishment of AF-MSCs from AF
Informed consent was obtained from all subjects, and all studies were conducted with strict adherence to guidelines of the Institutional Review Board of Korea University. Amniotic fluid (AF) was obtained by amniocentesis performed for fetal karyotyping between 16- and 20-week gestation. AF-derived cells were cultured from AF as previously described, or obtained from confluent back-up human amniocentesis cultures from a clinical cytogenetics laboratory [3]. Briefly, primary cell cultures were established in α-MEM medium (Grand Island, NY) containing 15% ES-FBS (ES qualified-fetal bovine serum), 1% glutamine, and 1% penicillin/streptomycin (Gibco/Invitrogen, Carlsbad, CA), supplemented with 18% Chang B and 2% Chang C (Irvine Scientific, Santa Ana, CA) at 37°C in a 5% CO2 atmosphere. At this stage, a mixture of 2 morphologically distinct groups attached and formed colonies. For selective culture of AF-MSCs, cells were harvested by trypsinization and plated in low-glucose DMEM medium (Gibco/Invitrogen) supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT) and 4 ng/mL basic fibroblast growth factor (FGF2; R&D Systems, Minneapolis, MN), and incubated as described previously. A morphologically homogeneous population of AF-MSCs was obtained after 2 rounds of subculture. These AF-MSCs were maintained in a humidified atmosphere in an incubator under 5% CO2 at 37°C. The clonal AF-MSCs were subcultured routinely at a dilution of 1:3 and were not permitted to progress beyond ∼70% confluency. Cells were then tested for genetic markers, cellular surface antigens, and differentiation potentials. As controls, human bone marrow-derived mesenchymal stem cells (BM-MSCs) were purchased and cultured according to the manufacturer's instructions (Poietics; BioWhittaker, Taufkirchen).
Preparation of conditioned medium (CM)
AF-MSCs were plated at a concentration of 5 × 104, 1 × 105, 2.5 × 105, or 5 × 105 cells/100-mm plate and incubated in proliferation medium overnight. The attached cells were washed 3 times with phosphate-buffered saline (PBS), and the medium was replaced with serum-free DMEM/F12 in order to generate CM that was serum-free and compatible for culture of AF-MSCs. The conditioned medium was prepared by incubating the cells for 3 days. The medium was then collected, centrifuged at 1,000 rpm for 5 min, and filtered through a 0.20-μm syringe filter. For in vivo experiments, the conditioned medium was further concentrated (5×) by ultrafiltration using centrifugal filter units with 5-kDa cutoff (Millipore Corporation, Bedford, MA) following the manufacturer's instructions.
Karyotype analysis
Karyotyping was performed by the Cytogenomic Services Facility of Samkwang Medical Laboratories. To analyze AF-MSC karyotype, AF-MSCs were cultured in proliferation medium as described above. Cell division was blocked in metaphase by the addition of 0.05 μg/mL colcemid (Gibco/Invitrogen) for 1–2 h [25,26]. The chromosomes were then visualized by G-band staining. At least 100 metaphase cells were analyzed, and a minimum of 10 were karyotyped for each line.
FACS analysis
FACS analysis of each sample was performed according to the protocol described previously [26,27]. Briefly, AF-MSCs and BM-MSCs were trypsinized and transferred into FACS tubes at a concentration of 1 × 106 cells/tube (BD Biosciences Clontech, Palo Alto, CA). After they were rinsed twice with cold buffer solution [DPBS with 1% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO), 0.02% sodium azide, at pH 7.4], the cells were incubated at 4°C for 1 h with primary antibody (Supplementary Table 1; Supplementary materials are available online at
Colony-forming unit fibroblast (CFU-F) assay
The CFU-F assay was performed by first plating AF-MSCs or BM-MSCs at a density of 100 numbers/well (6-well plates). After 14 days of culture, cells were washed twice with PBS and fixed with 10% formalin for 20 min at room temperature. To visualize and enumerate CFUs, the cells were stained with 0.01% crystal violet solution for 20 min at room temperature and then washed with deionized water and air dried. CFU-colonies were typically between 5 and 8 mm in diameter and were scored macroscopically [26,28,29].
Adipogenic differentiation
Differentiation of each sample was performed according to the previously described protocol [26,27]. Briefly, the cells were seeded at a density of 1.5 × 104 cells/cm2 in 6-well culture dishes (BD Biosciences, San Jose, CA) and cultured in high-glucose DMEM with 10% FBS until they reached 100% confluence. They were then subjected to 3 cycles of induction/maintenance by sequentially culturing the cells in adipogenic induction medium [high-glucose DMEM (Invitrogen, Carlsbad, CA) supplemented with 1 mM dexamethasone (Sigma-Aldrich, St. Louis, MO), 0.5 mM 3-isobutyl-1-methyl-xanthine (Sigma-Aldrich), 10 ng/mL recombinant human insulin (Sigma-Aldrich), 100 mM indomethacin (Sigma-Aldrich), and 10% FBS] for 7 days, adipogenic maintenance medium (high-glucose DMEM supplemented with 10 ng/mL recombinant human insulin and 10% FBS), and control medium (high-glucose DMEM supplemented with 10% FBS) for 7 days; this process was repeated 3 times. After differentiation, the cells were fixed with 10% formalin (Sigma-Aldrich), washed, and stained with 2% (wt/vol) Oil Red O reagent (Sigma-Aldrich) for 5 min at room temperature to examine the generation of oil droplets in the cytoplasm.
Osteogenic differentiation
Differentiation of each sample was performed according to the previously described protocol [26,27]. Briefly, cells were seeded at a density of 3 × 103 cells/cm2 in 6-well culture dishes (BD Biosciences), cultured in high-glucose DMEM (Gibco/Invitrogen) with 10% FBS until they reached 70%–80% confluence, and then fed twice a week for 2.5 weeks with osteogenic induction medium [IMDM basal medium (Gibco/Invitrogen) supplemented with 100 nM dexamethasone, 10 mM β-glycerophosphate, 0.2 mM ascorbate, and 10% FBS] and control medium [IMDM basal medium supplemented with 10% FBS]. Osteogenic differentiation was detected by fixing the cells with 10% formalin (Sigma-Aldrich) for 15 min at room temperature and then staining them with Alizarin red S [26,30].
Chondrogenic differentiation
To induce chondrogenic differentiation, cells were trypsinized, transferred into a 15-mL polypropylene tube, centrifuged at 1,000 rpm for 5 min to form a pelleted micromass at the bottom of the tube, and then treated with chondrogenic medium [high-glucose DMEM supplemented with 0.1 M dexamethasone, 50 g/mL AsA (Sigma-Aldrich), 100 g/mL sodium pyruvate (Sigma-Aldrich), 40 g/mL proline (Sigma-Aldrich), 10 ng/mL TGF-1 (R&D systems, Minneapolis, MN), and 50 mg/mL ITSpremix (Gibco/Invitrogen), 6.25 g/mL insulin, 6.25 g/mL transferrin (Sigma-Aldrich), 6.25 ng/mL selenious acid (Sigma-Aldrich), 1.25 mg/mL BSA, and 5.35 mg/mL linoleic acid (Sigma-Aldrich)] and control medium [high-glucose DMEM] [26,27]. Medium changes were carried out twice weekly and chondrogenesis was assessed at weekly intervals. After 2–4 weeks of culture, the cells were washed twice with PBS, fixed in 4% paraformaldehyde, and visualized either by staining with Alcian Blue (Sigma-Aldrich) or by immunofluorescence staining with Type II collagen antibody (Chemicon, Temecula, CA).
Immunofluorescence staining and quantification
Cultured undifferentiated and/or differentiated AF-MSCs were subjected to immunofluorescence staining, as previously described [26,31]. The primary antibodies used are listed in Supplementary Table 1. To determine the percentage of cells expressing a given marker protein, at least 3 fields in any given experiment were photographed and the number of positive cells relative to the total number of DAPI-labeled nuclei was determined.
RT-PCR and real time polymerase chain reaction
RNA was prepared from samples using TRIzol according to the manufacturer's instructions (Invitrogen), and cDNA was generated using Reverse Transcriptase II (Invitrogen) according to the manufacturer's instructions. To amplify various marker genes, 25 ng cDNA was used along with the PCR primers (Bioneer, Daejeon) under the conditions outlined in Supplementary Table 2. Reaction mixtures (20 pL) were prepared as described above. The amplification program consisted of 24–35 cycles of the following parameters: 94°C for 30 s; annealing at 62°C for 30 s, and extension at 72°C for 30 s, followed by a final amplification step for 10 min at 72°C. We confirmed that the levels of the different PCR targets generated by 24–35 PCR cycles were in the linear range [32]. Real-time RT-PCR was conducted using the iCycler IQ (Bio-Rad, Hercules, CA). Reactions were performed using SYBR-Green PCR Master Mix (Bio-Rad). As an internal control, levels of glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) were quantified in parallel with target genes. Normalization and fold changes were calculated using the ΔΔCt method [33].
In vitro proliferation assay
Cell growth was determined by plating fibroblasts at a density of 5 × 104 cells/well (24-well plate) and synchronizing them in proliferation medium [34]. Twelve hours later, AF-MSC-CMs [AF-MSCs were plated at a density of 5 × 104 CM (5), 1 × 105 CM (10), 2.5 × 105 CM (25), or 5 × 105 CM (50) cells/100-mm plate] or nonconditioned medium was added to triplicate wells and cultured for 72 h. Cells were then fixed with 10% formalin and stained with 0.01% crystal violet solution. Crystal violet solution from stained cells was extracted using 10% acetic acid and subjected to spectrophotometric analysis (600 nm) to determine relative cell growth rates.
Incorporation of BrdU
Cells were plates in 4-well plates. After serum starvation, cells were stimulated with CM in the BrdU (10 μM; Sigma-Aldrich). After 24 h, cells were fixed and stained for BrdU incorporation by immunostaining using BrdU staining kit (Invitrogen). The BrdU label index, defined as the proportion of total cells incorporating BrdU into the nucleus, was determined by counting BrdU-immunolabeled cells over total cells under phase contrast. The percentage of BrdU incorporation was calculated according to the following formula: % of BrdU-positive cells = (number of BrdU-positive cells/number of cells) × 100. The data from 3 independent experiments have been averaged, and the mean and standard deviation are shown [35,36].
Human cytokine protein array
A human cytokine antibody array (Panomics, Redwood City, CA) was used to identify the cytokines present in non-conditioned and AF-MSC-conditioned media (AF-MSC-CM). All assays were performed according to the manufacturer's instructions. Briefly, after blocking the nonspecific binding sites, membranes were incubated in nonconditioned medium or AF-MSC-CM overnight at 4°C. After membranes had been washed, a biotin-conjugated antibody was incubated for 16 h at 4°C. The membranes were again washed and then incubated with horseradish peroxidase–conjugated streptavidin at RT for 45 min. Finally, the membranes were incubated with detection buffer (ECL solution), and exposed to Hyperfilm.
Enzyme-linked immunosorbent assay (ELISA)
Quantification of cytokines in conditioned medium (AF-MSC-CM) was performed by ELISAs (Ray Biotech Inc., Norcross, GA) [37] according to the manufacturer's instructions. TGFβ, VEGF, Interleukin-8 (IL-8), and MMP3 were analyzed in AF-MSC-CM. The optimal density of the color reaction was detected at a wavelength of 450 nm using a chemiluminescence reader. The background signal detected at 450 nm was subtracted from the values. Delta values were normalized to the extinction obtained from standard curves, and protein contents were calculated for each sample. Cytokine amounts were normalized to identical numbers of cells per milliliter of medium.
Western blot analysis
To detect protein expression in response to treatment with AF-MSC-CM or nonconditioned medium, cells were plated at a density of 3 × 105 cells/100-mm plate, allowed to attach for 12 h, and cultured with serum-free DMEM for 48 h. Cells were then treated with AF-MSC-CMs or control medium for the indicated times (30 min–8 h). To detect protein expression and modification in response to treatment with the TGF-β inhibitor, SB505124 (Sigma-Aldrich), cells were plated at a density of 7 × 105 cells/100-mm plate and cultured for the indicated times (30 min) in AF-MSC-CM in the presence or absence of the inhibitor. Total protein was extracted with RIPA buffer. Lysates were centrifuged at 12,000g for 30 min at 4°C. Protein concentrations were determined using the Bradford assay kit (Bio-Rad, Hercules, CA). Proteins were separated using precast 4%–12% gradient SDS-PAGE (Invitrogen) and transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA). Blots were incubated with the indicated primary antibodies at 4°C and horseradish peroxidase–conjugated anti-mouse and anti-rabbit secondary antibodies (1:1,000 dilution) at RT. All primary antibodies were used at a final concentration of 1 μg/mL. Blots were then visualized using a chemiluminescence detection system according to the manufacturer's instructions (ECL kit; Pierce, Rockford, IL).
In vitro wound-healing assay
Wound-healing assays were performed as previously described [34,38]. Briefly, cells were seeded into 6-well dishes at a density of 7 × 105 cells/well. The dishes were cultured as confluent monolayers and then further synchronized in high-glucose DMEM containing 10% FBS at least for 5 h. The cells were scratched once per well with a 200-μL pipette tip to create an artificial wound. In vitro injury was induced by creating linear scratches of 500-μm wide strip of cells [34]. The scratch border was marked with a fine black line immediately after the scraping. Wounded cell cultures were then incubated in the presence of AF-MSC-CMs or control medium for 6–24 h. The migration of cells was assessed as a function of how far from the scratch line the cells had progressed and the overall number of cells migrating over the 24-h period.
In vivo wound healing assay
ICR mice (8 week-old, female, body weight 20 grams) were obtained from The Jackson Laboratory. The animals were randomly divided into three groups and the excisional wound splinting model was generated as described previously [39,40]. In brief, after hair removal from the dorsal surface and anesthesia, two 2-mm full thickness excisional skin wounds were created on each side of the midline. Each wound received either control medium or AF-MSC-CM by both subcutaneous injection around the wound and topical application on the wound bed. A donut-shaped silicone splint was positioned so that the wound was centered within the splint. An immediate-bonding adhesive (Krazy Glue) was used to fix the splint to the skin, followed by interrupted sutures to stabilize its position and the application of Tegaderm (3M) over the wounds. Digital photographs of the wounds were taken at 0, 4, 6, and 8 days. Time to wound closure was defined as the time at which the wound bed was completely re-epi-thelialized and filled with new tissue. The wound area was measured by tracing the wound margin and calculated using an image analysis program (NIH Image). The percentage of wound closure was calculated as: (area of original wound – area of actual wound)/area of original wound × 100. The percentage of AF-MSC-CM wound closure mean the value of DMEM/F12 converted to 100. Mice were sacrificed at 8 days, at which time skin samples, including the wound and 2 mm of the surrounding skin, were harvested for histology using a 2 mm punch biopsy. Tissue specimens were fixed in 10% freshly prepared formalin for 24 h and embedded in paraffin [34]. Four-micron-thick tissue sections were cut using a microtome and collected on superfrost plus slides. Tissue sections were then rehydrated by gradual immersion in 70, 80, 95, and 100% ethanol, cleared with xylene, and finally mounted in permount solution (Fisher Scientific, Springfield, NJ). Tissue section is washed with xylene to remove paraffin and immunohistochemical staining was performed on each fragment for p-Smad2 and Smad2. Primary antibodies against phospho-Smad2 and Smad2, which diluted at 1:100 ratio, were induced to show reactions at 4°C for overnight. The secondary Cy3-labeled antibodies were induced to show reaction for 2 hours and washed in TBST. Sections were washed again and mounted with Vectashied mounting medium containing 4,6-diamidino-2-phenylindole (DAPI). Picture was taken with Olympuse DP70 camera system.
Statistical analysis
All values are expressed as means ± SD. Student's paired t-test was performed for comparison of data of paired samples. A probability (P) value <0.01, 0.05 was considered significant.
Results
Establishment of clonal stem cell lines from amniotic fluid
Initially, we obtained AF-derived cells from either AF or back-up amniocentesis. Briefly, cells isolated from AF were cultured for 12–14 days at which point they reached confluency (Fig. 1A, a–d). However, the population was somewhat heterogeneous and consisted primarily of 2 types of cells: 1 type was similar to fibroblasts and the other was flat and circular, resembling epithelial cells. When the cells reached confluence, they were treated with trypsin and EDTA for 5 min, which resulted in the detachment of a group of cells from the bottom of the flasks. The cells were washed with PBS, and transferred into a new 60 mm tissue culture plate flask for the first passage culture as described in Materials and Methods. Two rounds of subculture were performed, and by the third passage, the cells had become morphologically homogeneous and consistently exhibited a fibroblast-like morphology until the end of the culture period (Fig. 1A, e). The third and subsequent passages were carried out in 100-mm flasks under the same conditions at a split ratio of 1:3. We succeeded in culturing AF-derived cells from every AF sample tested, and the results were similar regardless of the patient source (n = 50; data not shown). These established cell lines were designated as AF-MSCs and were used in all subsequent experiments.
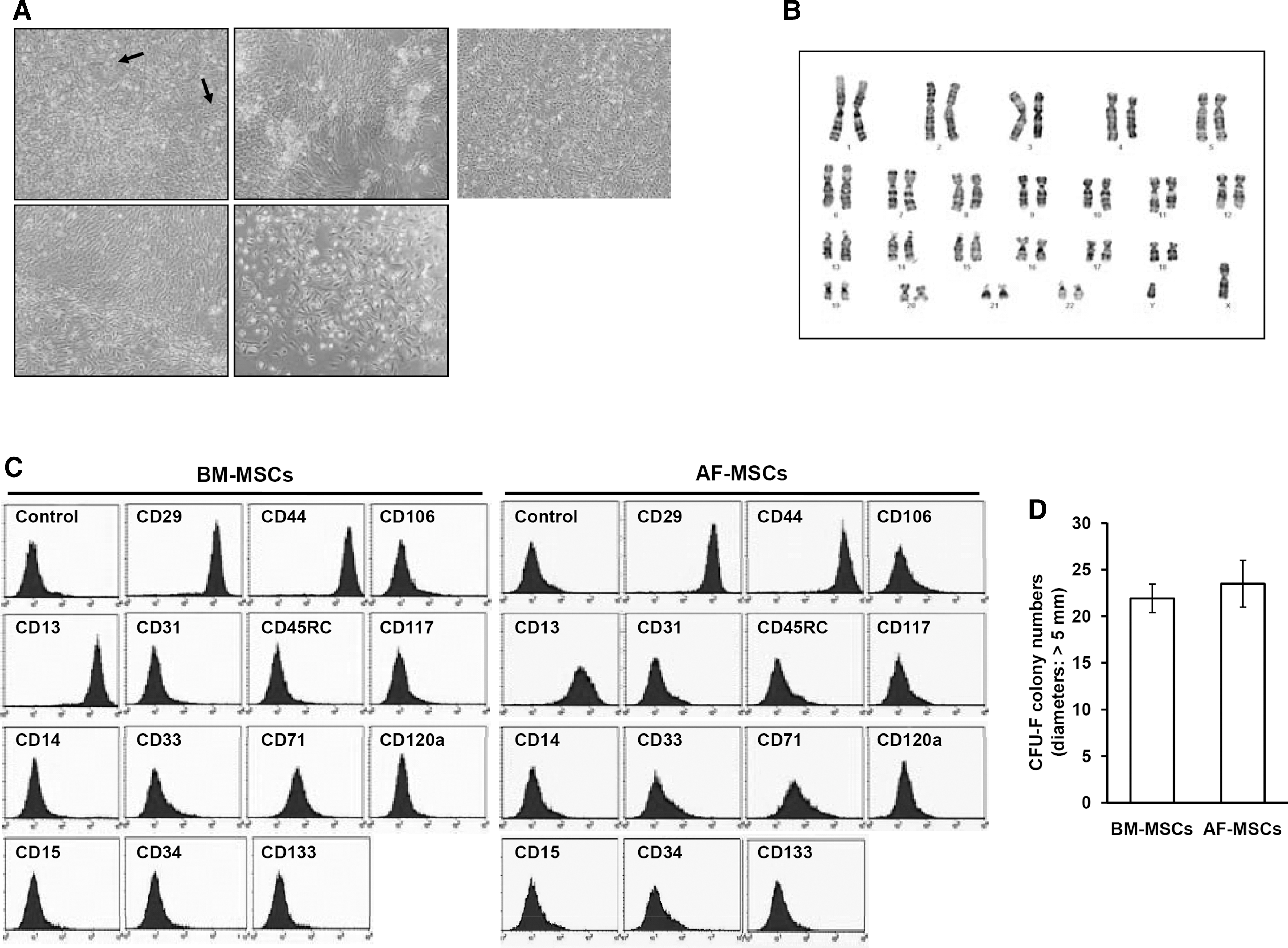
Characterization of isolated amniotic fluid derived-mesenchymal stem cells (AF-MSCs). (
Analysis of the proliferative capability of the cells over a 6-day culture period revealed that the cell population doubled every 31 ± 2 h (data not shown). Routinely, we maintained cells in culture for 17–25 passages in our laboratory, and G-banding analysis of AF-MSCs revealed a normal chromosomal karyotype (Fig. 1B), which indicates that these cells remain chromosomally stable over long-term culture. However, during in vitro culture, AF-MSCs possess a limited life span and undergo replicative senescence, indicated by loss of proliferation, and become more flattened and enlarged with the positive staining by SA-β-gal (data not shown).
FACS analysis using multiple surface epitopes demonstrated that >90% of AF-MSCs (passage 17) and bone marrow (BM)-MSCs (passage 3) expressed the typical MSC marker proteins CD13, CD29, and CD44. In contrast, known hematopoietic stem cell (HSC), neural stem cell (NSC), and endothelial cell markers (CD14, CD15, CD31, CD33, CD34, CD45, CD71, CD106, CD117, CD120a, and CD133) were not expressed [26,41,42] (Fig. 1C). The CFU-F assay provides a convenient means of assessing the proliferation and clonogenic capacity of cells expanded in culture. To compare the colony-forming ability of AF-MSCs with that of BM-MSCs, a CFU-F assay was conducted and the colonies with a diameter >5 mm were counted. The CFU-F capacity of AF-MSCs was similar to that of BM-MSCs. The results showed that over 14 days, 23.5 ± 2.51 colonies formed from BM-MSCs plated at 100 cells/cm2, whereas 21.93 ± 1.53 colonies formed from AF-MSCs (Fig. 1D).
Mesenchymal differentiation potential of AF-MSCs
To determine whether AF-MSCs could differentiate into various mesenchymal lineage cells, we examined the differentiation of AF-MSCs into osteoblasts, adipocytes, and chrondrocytes [26]. The AF-MSCs from each of the 5 patients differentiated with similar efficiencies into all the 3 cell types, as described below.
To examine whether the AF-MSCs could differentiate into adipocytes, the AF-MSCs and BM-MSCs (used as comparative control for this study) were subjected to 3 cycles of culture in adipogenic induction and maintenance media (AS+) or control media (AS−) [26,27]. After 2 weeks, only the differentiated AF-MSCs and BM-MSCs exhibited morphological changes and small neutral oil droplets in the cytoplasm, revealed by Oil Red O staining, which started appearing within 1 week of adipogenic induction (Fig. 2A). Moreover, RT-PCR analysis of adipogenic gene expression revealed upregulation of aP2 (adipocyte fatty acid–binding protein 2), PPARr2 (peroxisome proliferator–activated receptor γ2), and LPL (lipoprotein lipase; Fig. 2B), demonstrating that AF-MSCs, similar to BM-MSCs, are indeed capable of differentiation into adipogenic lineages.

Multilineage differentiation capacity of amniotic fluid derived-mesenchymal stem cells (AF-MSCs). Cells at passages 5–7 were analyzed for their ability to differentiate in vitro into adipocyte-like (
To examine whether AF-MSCs were capable of osteogenic lineage differentiation, AF-MSCs and BM-MSCs were cultured in osteogenic induction medium (OS+) or control medium (OS−) in 6-well plates for 2–3 weeks. Alizarin red staining revealed that differentiated AF-MSCs and BM-MSCs developed a calcium-rich mineralized matrix along the cell membrane that appeared as large red aggregates embedded in the ECM (Fig. 2C). RT-PCR analysis of osteogenic gene expression revealed upregulation of osteopontin and osteocalcin (Fig. 2D), demonstrating that AF-MSCs, similar to BM-MSCs, are indeed capable of differentiation into osteogenic lineages.
To examine whether AF-MSCs could differentiate into chondrogenic lineages, AF-MSCs and BM-MSCs were induced to form a sphere by centrifuging 5 × 105 to 5 × 106 cells in a 15-mL polypropylene tube and culturing this sphere in either growth medium (CS−) or chondrogenic induction medium (CS+). After 3 weeks, only differentiated cells showed evidence of acidic mucopolysaccharides in both, as revealed by Alcian Blue staining, and immunofluorescent staining of Type II collagen (Fig. 2E). RT-PCR analysis of chondrogenic gene expression showed upregulation of aggrecan, and Type II collagen (Fig. 2F), demonstrating that AF-MSCs, similar to BM-MSCs, are capable of differentiation into chondrogenic lineages.
Stimulatory effect of AF-MSC-derived conditioned medium (AF-MSC-CM) on the proliferation of dermal fibroblasts
Because the extensive proliferation of fibroblasts is closely linked to the wound-healing process [19,43], we examined the effects of AF-MSC-CM on the proliferation of dermal fibroblasts. We first established the initial plating density of AF-MSCs for the optimal production of AF-MSC-CM. AF-MSCs were plated in DMEM culture medium at a density of 5 × 104, 1 × 105, 2.5 × 105, and 5 × 105/100-mm plate. One week later, cells were trypsinized and counted (data not shown). The optimal plating density was determined to be 2.5 × 105 cells/100-mm plate, followed by densities of 1 × 105, 5 × 105, and 5 × 104 cells/100-mm plate (data not shown). These data suggest that the initial plating density of AF-MSCs has a significant impact on their proliferation potential, whereas the phenotype of the cells (colony-forming ability and in vitro multilineage differentiation potential) remained unaffected by plating density (data not shown).
We next tested the effect of AF-MSC-CM on the proliferation of dermal fibroblasts. First, we examined the effect of AF-MSC-CM produced from differentially plated AF-MSCs. The number of viable dermal fibroblasts was significantly increased when incubated with AF-MSC-CM from any plating density compared to basal culture medium (Fig. 3A). We then tested whether the increased proliferation of dermal fibroblasts resulted from increased cell-cycle progression. To determine whether cells that entered S phase after AF-MSC-CMs were able to progress completely through the cell cycle, cells were labeled with BrdU during 24 h of culture, and then the number of BrdU-positive cells was counted at 24 h. The absolute number of BrdU-positive cells was higher in the CM-treated cells compared with control (DMEM/F12-treated cells; Fig. 3B). In order to investigate the molecular mechanism that mediates cell-cycle progression in dermal fibroblasts after treatment with AF-MSC-CM, we examined whether AF-MSC-CM could stimulate steady-state mRNA levels of cell cycle–related genes. Real-time PCR analysis demonstrated that AF-MSC-CM enhanced the expression of Cyclin A and Cyclin E (Fig. 3C).

Effects of amniotic fluid derived-mesenchymal stem cells with conditioned media (AF-MSC-CMs) on the proliferation of dermal fibroblasts. (
Antibody-based protein array analysis of secreted proteins from AF-MSCs
Mesenchymal stem cells release a large number of growth factors and cytokines that are important for the repair of injured tissues [44 –46]. To examine whether AF-MSCs also secrete growth factors and cytokines essential for the repair of wounded tissue, we performed antibody-based protein and ELISA array analyses of AF-MSC-CM and non-CM (DMEM/F12). Compared to non-CM, AF-MSC-CM contained dramatically increased levels of several cytokines, including VEGF, TGF-β IL-8, IL-6, TNF-α, IL-4, macrophage inflammatory protein (MIP)-4, Apol/Fas, and Leptin (Fig. 4A). These molecules are known to be important in normal wound healing [47]. Further analysis of the amounts of IL-8, VEGF, TGF-β, and MMP3 in AF-MSC-CM or non-CM by ELISA confirmed that they were increased in AF-MSC-CM (Fig. 4B). These results suggest that AF-MSCs secrete high levels of growth factors and cytokines that are important for wound healing.
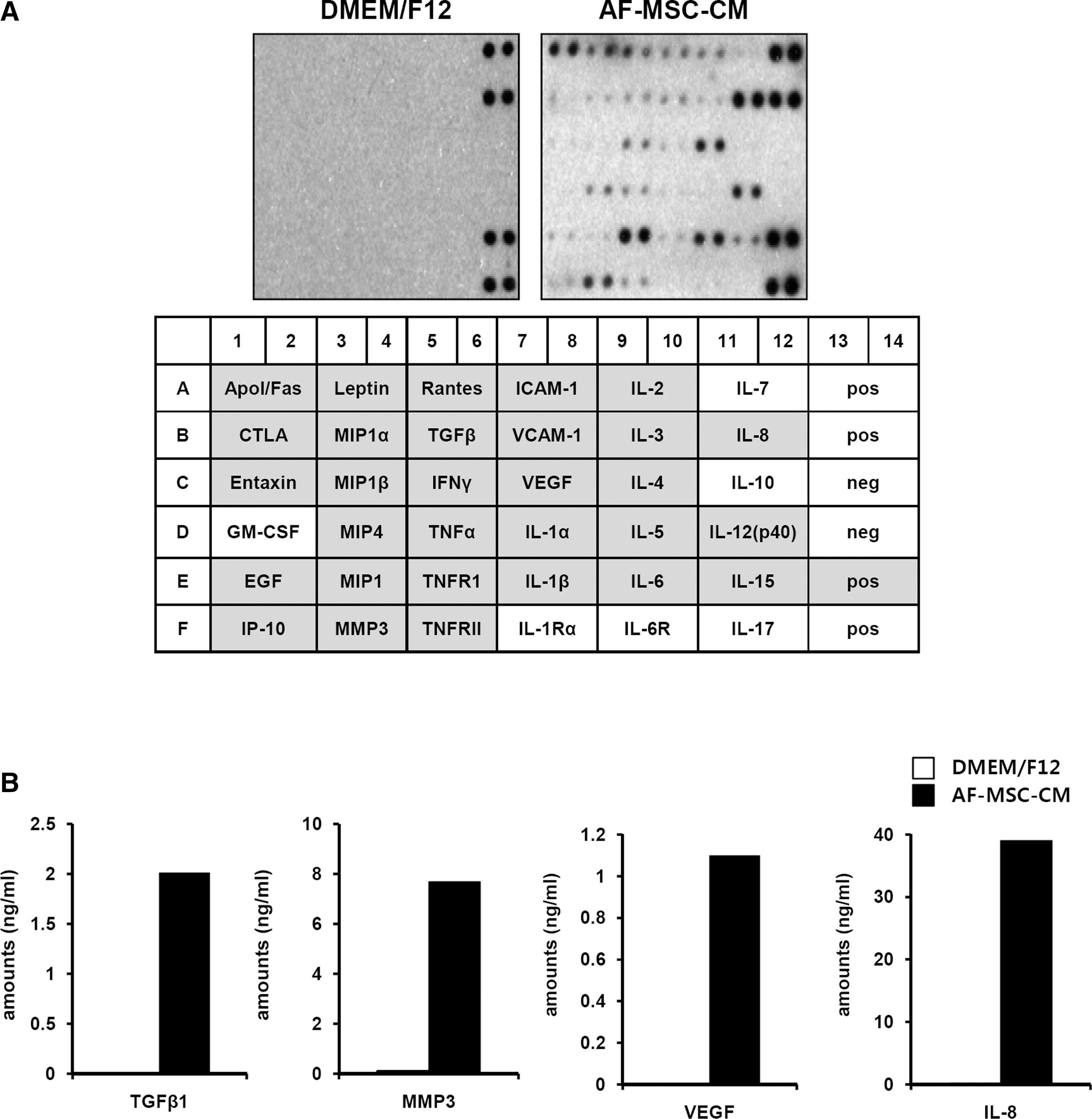
Protein levels of cytokines in amniotic fluid derived-mesenchymal stem cells with conditioned media (AF-MSC-CMs). (
Stimulatory effect of AF-MSC-CMs on the migration of dermal fibroblasts
Wound healing requires recruitment of fibroblasts into the wound from surrounding tissues and subsequent proliferation of these cells. Because previous experiments had demonstrated that MSC-CM exhibited biological effects relevant to the migration of dermal fibroblasts [48], we investigated whether AF-MSC-CM induced dermal fibroblast migration. Confluent plates of dermal fibroblasts were denuded in a straight line with a 200-μL pipette tip to create an artificial wound, using the standard “scratch test” technique [34,38]. The cultures were then incubated with AF-MSC-CM, and plates were imaged by phase-contrast microscopy at the 6-h end point to assess the degree of cell migration from the original line (Fig. 4A; pictures). Quantitation of migrated cells revealed a significant increase in the migratory capacity of dermal fibroblasts when cells were incubated with various AF-MSC-CMs compared with control medium (Fig. 4A; graphs). Fibroblasts incubated in AF-MSC-CM obtained from cells plated at 2.5 × 105 migrated faster than their counterparts. However, the proliferation and migration effects of all of the AF-MSC-CMs were completely abolished by pre-treatment of the media with heat (data not shown).
Injury to the skin resulting in a wound is known to trigger a repair process that is characterized by major alterations in both the composition and structure of the ECM [49]. In order to assess the effect of AF-MSC-CM on dermal fibroblasts in an artificial wound model, real-time PCR was performed to determine changes in the mRNA expression levels of ECM molecules involved in wound healing. Compared to control media, fibroblasts incubated in AF-MSC-CM expressed significantly greater amounts of ECM molecules, including Collagen type III, vitronectin, fibronectin, MMP1, Syndecan 2, Syndecan 4, SPP 1, and Elastin (Fig. 5). Moreover, fibroblasts incubated in AF-MSC-CM obtained from cells plated at a density of 2.5 × 105 expressed considerably greater amounts of ECM molecules than their counterparts. Thus, these findings indicate that AF-MSC-CM efficiently enhances wound healing-related events, such as collagen production, fibroblast outgrowth, and cell migration in dermal fibroblasts.
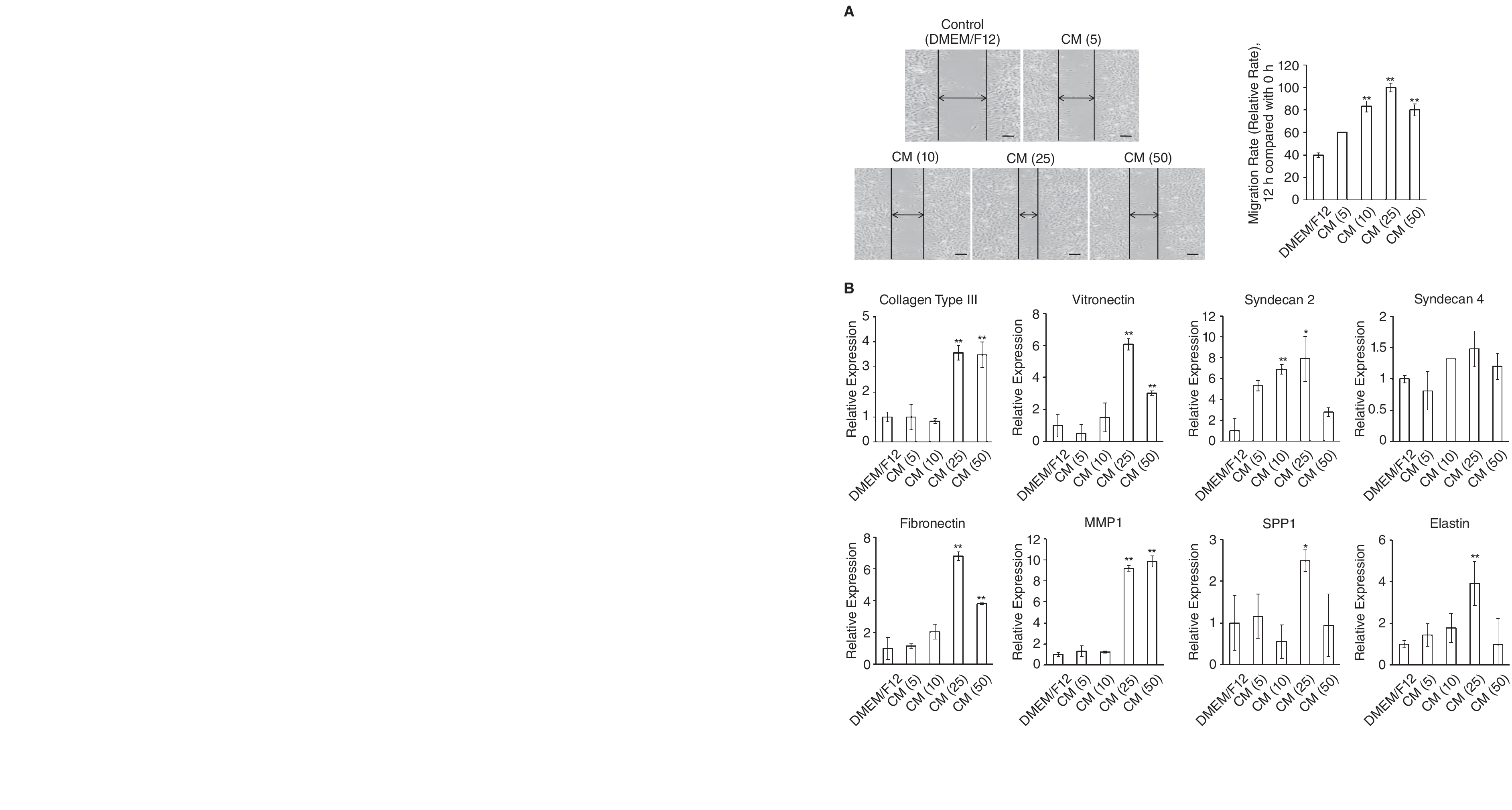
Effect of amniotic fluid derived-mesenchymal stem cells with conditioned media (AF-MSC-CMs) on dermal fibroblast migration. (
AF-MSC-CM regulates the SMAD2 pathway in dermal fibroblasts
The above results prompted us to examine the molecular mechanisms by which AF-MSC-CM enhances the proliferation and migration of dermal fibroblasts. Previous studies have reported that activation of TGF-β in dermal fibroblasts plays an important role in wound healing [24]. Therefore, we used western blotting to compare the phosphorylation levels of TGF-β/SMAD signaling pathways in dermal fibroblasts treated with non-CM or AF-MSC-CM. As shown in Figure 6A, compared to those treated with non-CM, dermal fibroblasts treated with AF-MSC-CM expressed higher levels of phosphorylated SMAD2, but not phosphorylated SMAD3. Upregulation of SMAD2 phosphorylation is consistent with previous data indicating that SMAD3 may be the principal mediator of keratinocyte growth inhibition, as well as of chemotaxis, whereas SMAD2 principally mediates ECM responses in fibroblasts [50].

Amniotic fluid derived-mesenchymal stem cells with conditioned media (AF-MSC-CMs) regulates the TGFβ/SMAD pathways in dermal fibroblasts. (
We next tested whether inhibition of the TGF-β pathway would directly suppress migration of dermal fibroblasts treated with AF-MSC-CM. SB505124 is a small molecule inhibitor identified on the basis of its ability to inhibit the in vitro kinase activity of ALK5. Smad2 is activated by ALK4, ALK5, and ALK7. SB505124 inhibited the TGF-induced phosphorylation of Smad2 [51]. Treatment with the TGF-β receptor inhibitor, SB505124, resulted in a significant reduction of phosphorylated SMAD2 (Fig. 6B). Further, the addition of SB505124 to AF-MSC-CM strongly suppressed dermal fibroblast migration into scratch wounds (Fig. 6C). Quantitation of migrated cells revealed a significant decrease in the migratory capacity of dermal fibroblasts when they were incubated with SB505124 in AF-MSC-CM (Fig. 6D). Together, these data demonstrate that TGF-β signaling is an important mediator of the migration of dermal fibroblast induced by AF-MSC-CM.
AF-MSC-CM enhances wound healing in vivo
To examine whether AF-MSC-released factors could enhance wound healing, we both injected and topically applied 100 μL of concentrated AF-MSC-CM to excisional wounds created in ICR mice. Similar wounds treated with vehicle medium (non-CM) were used as controls. Careful measurements of the wounds at 6 days indicated that AF-MSC-CM significantly accelerated wound closure compared to vehicle control medium (Fig. 7A). In addition, the beginning of re-epithelialization from the wound edge was clearly observed in the wounds treated with AF-MSC-CM. The degree of wound closure in control and AF-MSC-CM-treated mice was quantitatively measured in Figure 7B. H&E staining of wounded skins was performed at 20 days after surgery; however, no significant difference in skin structure was observed between control and AF-MSC-CM-treated wounds (Fig. 7C). These data suggest that AF-MSC-CM accelerates the normal wound-healing process without any abnormal effects, such as granulation or epidermal hyperplasia. We performed immunostaining to show the activation of smad pathway in vivo. We confirmed that Smad2 was detected both DMEM/F12-treated group and CM-treated group, while pSmad2 was detected only in CM-treated group. These results have shown that AF-MSC-CM had wound-healing effect by Smad pathway in vitro and in vivo.
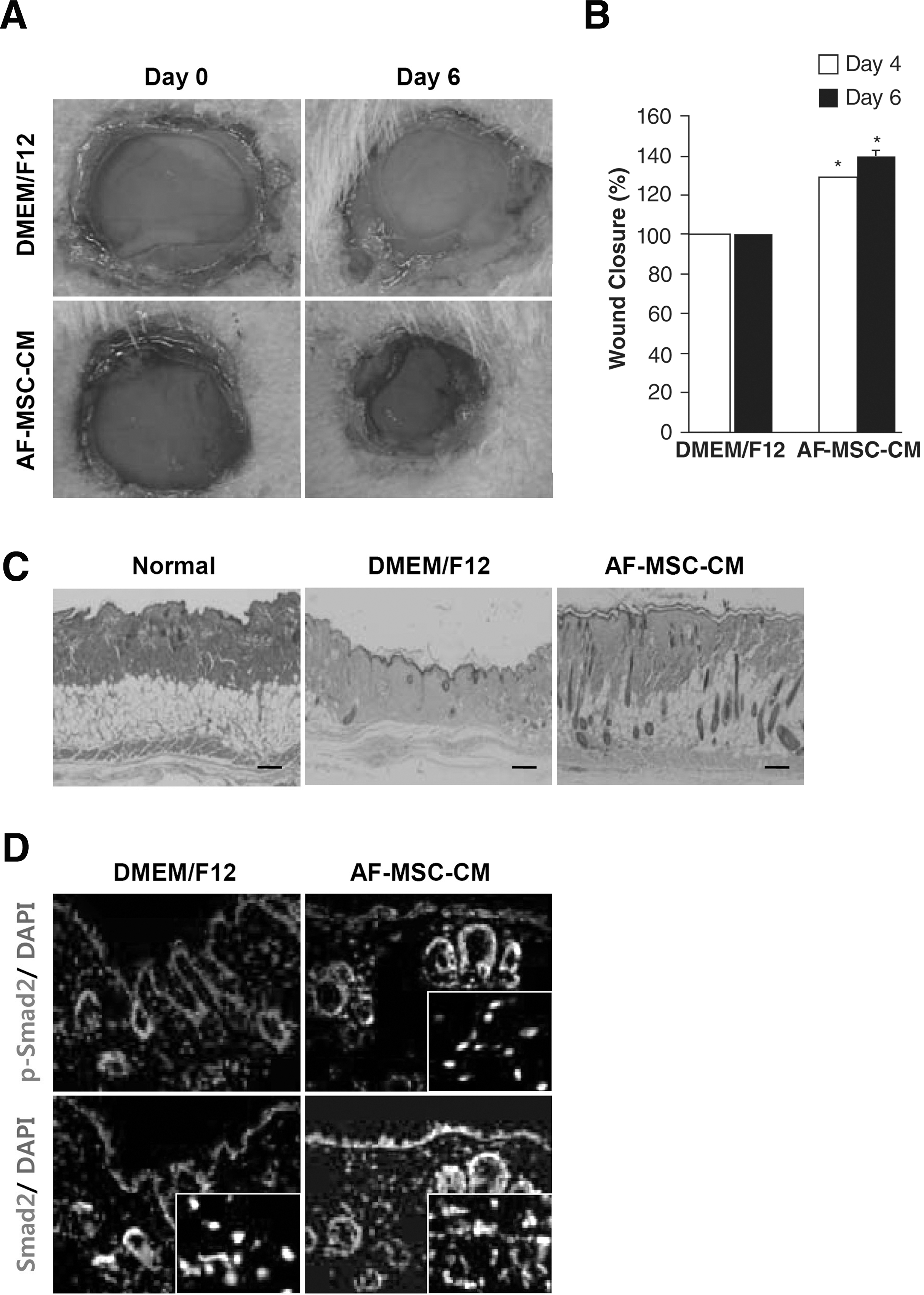
Effect of AF-MSC-CMs on wound closure. (
Discussion
Chronic wounds are a common affliction and often difficult to treat. In spite of recent advances from breakthroughs in growth factor research and the development of bioengineered skin, up to 50% of chronic wounds that have been present for more than a year remain resistant to treatment [52]. Therefore, novel treatments to improve the effectiveness of chronic wound closure and epithelial regeneration are required. One of the most promising interventions is the use of embryonic stem cells (ESCs) and adult stem cells for wound repair and regeneration of injured tissues. ESCs have a great capacity for self-renewal and plasticity, but their use is limited by political and ethical considerations. The use of adult stem cells, especially MSCs from bone marrow and other tissues, such as adipose, nerve, and umbilical cord blood, is not subject to these concerns. Here, we have demonstrated that it is possible to isolate and extensively expand MSCs from human AF, and that AF-MSC-CM promotes proliferation, migration, and wound healing by dermal fibroblasts. AF stem cells have a self-renewal capacity, express embryonic cell lineage markers after culture in vitro, and share the same properties and differentiation capabilities as embryonic stem cells [53]. AF is an abundant source of fetal MSCs that exhibits a phenotype and multi-lineage differentiation potential similar to that of postnatal BM-derived MSCs [5]. We propose that AF is an attractive source of MSCs for use in wound repair and regeneration of injured tissue.
AF stem cells represent a heterogeneous population composed of 3 groups of adherent cells defined by their morphological aspects and growth characteristics. These groups are epithelioid E-type cells, amniotic fluid–specific AF-type cells, and fibroblastic F-type cells [54,55]. The fibroblastic F-type cells are believed to originate from mesenchymal tissue and usually appear late during routine AF cell cultivation [55]. The AF-MSCs isolated by our culture method are fibroblastic F-type cells in the AF, and we confirmed their mesenchymal origin. The AF-MSCs can be expanded rapidly and maintain their capacity to differentiate into multiple mesodermal cell types in vitro. AF-MSCs possess phenotypic characteristics similar to MSCs derived from other sources, such as term umbilical cord blood and bone marrow, and are positive for CD13, CD29, and CD44, and negative for CD14, CD31, CD34, CD33, CD45, and CD117 [56 –58].
Bone marrow–derived MSCs secrete a large number of growth factors and cytokines that are critical for the repair of injured tissues [44 –46]. In a previous study, implantation of BM-MSCs was found to improve wound healing associated with increased angiogenesis in normal and diabetic mice [59]. However, BM-MSCs were observed adjacent to the vasculature but not in the vascular wall, implying that BM-MSCs exert a paracrine effect in wound healing and angiogenesis. Furthermore, the concept of using MSC-CM for wound healing is supported by showing that these cells can be successfully used as a restorative therapy for the treatment of stroke, myocardial infarction, and osteogenesis imperfecta [60,61]. In this study, we showed that AF-MSC-CM contains high levels of growth factors, cytokines, and chemokines, and enhances wound healing by dermal fibroblasts in vitro and in vivo, implying a critical role for paracrine factors in AF-MSC-enhanced wound healing. Our examination of the secreted soluble factors in AF-MSC-CM revealed that AF-MSCs produce growth factors, cytokines, and chemokines such as TNF-α, VEGF, TGF-β, Leptin, IL8, IL-6, and TNFα, all of which are upregulated during the inflammatory phase of wound healing [43]. IL-6 is primarily produced by neutrophils and monocytes, and has been shown to be important for initiating the healing response. Its expression is increased after wounding and tends to persist in older wounds [62]. TNFα can induce the production of FGF7, suggesting that it can indirectly promote re-epi-thelialization [63]. IL-8 is produced by epithelial and fibroblast cells, and plays an important role in inflammation and wound healing [64]. Furthermore, IL-8 is a chemotactic stimulus for fibroblasts and accelerates their migration. It can also stimulate deposition of fibronectin and collagen during wound healing in vivo [65]. The production of these soluble mediators by AF-MSCs would be expected to complement endogenously produced growth factors and cytokines that regulate cellular processes such as chemotaxis, cell proliferation, cell signaling, ECM formation, and angiogenesis in the wound. In our study, AF-MSC-CM significantly improved the proliferation and migration of dermal fibroblasts, and enhanced the wound-healing process in a skin injury model. These results suggest that AF-MSCs secrete high levels of growth factors and cytokines valuable to normal wound healing, and that these cytokines, in combination with ECM molecules and/or other unknown factors, might account for the effect of AF-MSC-CM.
During wound healing, TGF-β plays an important role in inflammation, angiogenesis, re-epithelialization, and connective tissue regeneration. TGF-β expression levels have been shown to increase at the onset of injury [66], and it is involved in upregulation of the angiogenic growth factor, VEGF [67]. In addition, in vitro studies show that TGF-β plays a role in wound contraction by facilitating fibroblast contraction of the collagen matrix [68]. TGF-β signaling is dependent on the activation of SMAD2 and SMAD3 by heteromeric complexes of ligand-specific receptors. SMAD3 signaling inhibits the rate of wound healing by inhibiting keratinocyte proliferation and monocyte function, whereas SMAD2 signaling promotes healing by stimulating matrix production by fibroblasts [50]. Mice lacking SMAD3 exhibit accelerated wound healing, whereas increased SMAD2 stimulates keratinocyte migration and re-epithelialization during wound healing [69]. These results are in agreement with our finding that dermal fibroblasts treated with AF-MSC-CM expressed higher levels of phosphorylated SMAD2, but not SMAD3. In addition, treatment with a TGF-β receptor inhibitor decreased phosphorylated levels of SMAD2 and inhibited dermal fibroblast migration into scratch wounds. These results suggest that the increased levels of TGF-β in AF-MSC-CM may enhance the proliferation and migration of dermal fibroblasts during normal wound healing via the SMAD2 pathway. Although inhibition of TGF-β/SMAD2 pathway decreased the migration of fibroblast, inhibition rate was under 50%. Even though we have found that ERK and STAT3 [34,70] signaling pathways, which were related to wound healing, had no effect on wound healing (data not shown), we cannot exclude the possibility that both SMAD2 pathway and other growth factors could influence wound healing because AF-MSC secreted various cytokines, growth factors, chemokines, etc.
In summary, our data showed that AF-MSCs secreted high levels of cytokines, growth factors, and chemokines, which could enhance wound healing, and that AF-MSC-CM may represent a novel therapy to improve the effectiveness of tissue repair. Furthermore, considering their clinical efficacy and safety, AF-MSCs and their secretion factors have great potential for applications in cosmetic dermatology, especially in the treatment or regeneration of wounded skin.
Footnotes
Acknowledgments
This research was supported by a grant (SC-5150) from the Stem Cell Research Center of the 21st Century Frontier Research Program, funded by the Ministry of Education, Science and Technology, Republic of Korea; a grant from Stemmedience, Republic of Korea; a grant from the second phase of Brain Korea 21 project, Republic of Korea; and a Korea University Grant, Republic of Korea.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.