
Abstract
Cancer cells are unequal in a tumor mass and in established cultures. This is attributable to cancer stem cells with the unique ability to self-renew and to generate differentiating progeny. This ability is controlled at the level of asymmetric division by mechanisms that are yet not well defined. We found that normal and cancer keratinocyte fate was linked to the asymmetric distribution of epidermal growth factor receptor (EGFR) during mitosis. Although essential for epithelial cell proliferation, differentiation, and survival, this receptor was not present on the surface of cells satisfying criteria for stem cells such as quiescence, competence to produce functionally distinct daughters, high proliferative and clonogenic potential, sphere formation ability, and expression of stem cell markers. In contrast, keratinocytes displaying EGFR acquired a more differentiated phenotype, suggesting that EGFR may be involved in a switch from stem to transient amplifying cell fate. This switch was associated with changes in the expression profile of cell cycle, survival, and mitochondria controlling proteins that varied between normal and cancer cells. In conclusion, it appears that an unequal distribution of EGFR at mitosis controls keratinocyte fate by balancing quiescence and cycling of EGFR− cells, clearly malfunctioning in cancer. We believe that our findings provide mechanistic insights into the development of resistance to anti-EGFR therapies.
Introduction
The cellular homeostasis of any adult tissue is maintained by a small subpopulation of undifferentiated and long living stem cells. These cells divide asymmetrically to reproduce self and to generate committed progenitor cells that are able to replace terminally differentiated, lost, and damaged cells of the tissue [1]. In addition to this fundamental role in tissue homeostasis and a contrario substantiating it, numerous findings reveal aberrant maintenance and function of stem cells in cancers [2 –5]. Indeed, accumulating data provide evidence that development of hematopoietic and solid tumors relies on a small population of cancer cells known as cancer stem cells or tumor-initiating cells. These cells have the capacity to reinitiate tumor growth and recapitulate the cellular heterogeneity of the original tumor mass as a result of their self-renewing and differentiating abilities [6]. In normal tissue, these dual abilities, unique to stem cells, are functionally uncoupled at the level of cell division by mechanisms controlling the production of 2 uneven cells and involving unequal distribution of molecular determinants of cell fate, cell polarity, mitotic spindle orientation, and, as recently documented, unequal distribution of surface receptors [2,7]. Along this line, unequal abundance of surface epidermal growth factor receptor (EGFR) was shown to specify asymmetry in cell fate of a subset of dividing embryonic murine cortical cells in response to environmental signals [8].
EGFR is a member of the ErbB family expressed in neuronal, epithelial, and mesenchymal cells where it plays critical roles in proliferation, differentiation, and apoptosis [9 –11]. In the epidermis, EGFR is responsible for maintaining the epidermal phenotype. Its strong expression is confined to the basal cell layer of proliferating keratinocytes and diminishes progressively in suprabasal layers upon advancing differentiation [11 –14]. Not all keratinocytes with lower levels of cell surface EGFR belong to the differentiation cell compartment. Fortunel et al. [14] demonstrated that in the native epidermis a subpopulation of these cells was endowed with the stem cell abilities to self-renew and produce differentiating progeny. However, EGFR is frequently overexpressed in epithelial tumors and its constitutive expression is associated with poor prognosis [11,15], implying that EGFR+ rather than EGFR− cells have tumorigenic potential. Since, according to the cancer stem cell concept of tumorigenesis [16,17], the probability that all cancer cells have stem cell capacity is relatively low, we question whether cultures of cancer cells overexpressing surface EGFR contain a scarce subpopulation of cells devoid of surface EGFR and whether this would direct cell fate choices in cancer. We found EGFR− cells in the A431 squamous cell carcinoma cell line and provide evidence that these cells function as self-renewing cancer stem cells producing EGFR+ progeny that progressively dominate the cell culture. Considering the importance of EGFR as a target for therapeutic intervention in epithelial tumors, our demonstration that EGFR− cells have traits of cancer stem cells should be taken into consideration since these cells may be the origin of therapy resistance observed in clinical settings.
Materials and Methods
Cell culture
Squamous cell carcinoma (SCC) A431 cell line [18] was maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Gibco®, Invitrogen, France) in a humidified 5% CO2 incubator at 37°C. Primary cultures of SCC, basal cell carcinoma (BCC), and breast skin normal human keratinocytes (NHK) were generated from 10 discarded surgical specimens as described previously [19,20]. Storage and use of human biological samples were declared and performed according to ethical rules approved by the Department of Health, France. These latest cells (passage 0 and/or P1) were maintained in defined K-SFM supplemented with 1% penicillin/streptomycin (Gibco®, Invitrogen, France). To visualize EGFR, either 10 µg/mL of Alexa 488-coupled EGF (Molecular Probes, Eugene, OR) was added to live cells and images were acquired using Time Lapse AS MDW (Leica®) video microscopy or cells were stained with FITC-conjugated anti-EGFR antibody, fixed, and photographed using a DMR microscope (Leica®). To assess colony-forming units (CFU), sorted or unsorted cells were plated onto 35-cm plates at various (100–30,000 cells/plate) densities. Three weeks later, colonies were isolated using glass cloning rings or counted after staining with crystal violet. A minimum of 32-cell aggregates were considered as a small colony (paraclone) and cell aggregations >3 mm in diameter as a large colony (holoclone). An A431-GFP cell line expressing traceable green fluorescent protein (GFP) was created by injecting subcutaneously into 6-week-old SCID mice 106 per mouse of A431 cells stably expressing GFP. The A431-GFP stable transfectants were created in our lab by selecting and amplifying a single clone of neomycin (600 µg/mL)-resistant transfectants bearing the pcDNA3-GFP construct. For the injection, only FACS-sorted GFP+ cells were used. Six weeks later, the injected A431-GFP cells generated tumors that were recovered to make cell suspensions. Tumor cells were plated and A431-GFP+ cells were resorted according to their cell surface EGFR and GFP content and replated. To generate primary spheres, 2 × 104 cells were plated on ultra low adhesion plates (Corning Costar Co, ATGC, France) and cultured in DMEM/F12 medium supplemented with 20 ng/mL EGF (Stem Cells Biotechnologies, Vancouver, BC), 1× B27-supplement (Gibco®, Invitrogen, France), and 20 ng/mL rHu bFGF (PromoKine-PromoCell GmbH, Germany) in a humidified 5% CO2 incubator at 37°C for 10 days. Spheres were dissociated by a brief incubation with trypsin–EDTA solution (Gibco®, Invitrogen, France) and replated at a density of 2 × 104 cells/6-well low adhesion plate. Spheres (bigger than ∼50 cells) were counted under the microscope by 2 independent experimentators. Colony-forming unit (CFU) and sphere-forming unit (SFU) potential (%) were estimated according to the formula: number of colonies or spheres/number of plated live cells × 100.
Flow cytometry and cell sorting
The single cell suspension (4 × 105 cells/100 µL) of NHK (passage 0 or 1) and A431 cells were incubated in growth medium with phycoerythrin (PE)-conjugated anti-human EGFR antibody (Abcam, Paris, France) alone or in combination with fluorescein isothiocyanate (FITC)-conjugated anti-CD44 or CD95 antibodies (Immunotools, Germany) for 30 min at 37°C. Antibodies were used at a 1:50 dilution. After incubation, cells were rinsed with growth medium, centrifuged, resuspended in 500 µL of medium, and placed on ice before being analyzed by flow cytometry. Dead or apoptotic cells were excluded according to morphology and propidium iodide (PI; 2 µg/mL of Sigma Aldrich, France) labeling preceding acquisition. Acquisition was done on an EPICS XL-4 MCL flow cytometer (Beckman Coulter) and analyzed using Expo32 software. FITC, PE, and PI fluorescence intensities were recorded on FL1, 2, and 3 channels, respectively. Quadrants were determined based on the negative control staining with a corresponding isotype antibody.
Cell cycle analysis. Suspension of non-sorted cells was stained with FITC-conjugated anti-EGFR (Santa Cruz Biotechnology, Germany) antibody for 30 min at 37°C in darkness, then rinsed twice with PBS, fixed with ethanol 70%, and incubated at least 1 h at −20°C. After rinsing 3 times with cold PBS, cells were incubated with 50 µg/mL PI and 5 µg/mL RNase in PBS for 20 min at 37°C. Reaction was stopped by placing cells onto ice just before flow cytometry analysis.
Rhodamine123 (Rh123) exclusion assay. Cell suspension of 106 cells/mL in DMEM/10% FBS was loaded with 0.1 µg/mL Rh123 (1 mg/mL stock dissolved in DMSO) for 20 min at 37°C, rinsed twice, and resuspended in 0.5 mL of medium. Cells were placed at 37°C for an additional 60 min. After rinsing, cells were stained with PE-conjugated EGFR antibody for 30 min at 4°C. Two µg/mL PI was added just prior to flow cytometric analysis to discriminate dead cells. A minimum of 10,000 live cells was acquired. Rh123 green and PE and PI red fluorescence was recorded on FL1, FL2, and FL3 channels, respectively. Cytometric data were analyzed with Expo32 software. Cell sorting was carried out on either autoMACS Miltenyi Biotech columns according to included specifications and/or on an EPICS-ALTRA sorter (Beckman Coulter). After exclusion of debris and cell doublets, collection gates were set according to the negative control containing either unstained cells and stained with irrelevant isotype matching antibody or cells devoid of fluorescent dye. EGFR-labeled cells with fluorescence intensity equal to the negative control were considered as the EGFR− subpopulation, the remaining constituted the EGFR+ compartment. Collected subpopulations of cells were centrifuged, rinsed, and replated either for resorting or for sphere generation, CFU analysis, and immunohistochemistry.
Immunofluorescence
Paraffin-embedded sections of human epidermis were deparaffinized, rehydrated with PBS, and saturated 30 min in PBS supplemented with 1% BSA plus 1% goat serum. Then monoclonal FITC-conjugated anti-EGFR antibodies (Santa Cruz Biotechnology, Germany) were added (dilution 1/50) and slides were incubated for 2 h at room temperature in PBS/1% BSA/1% goat serum. For cell slides, cell cytospins were permeabilized with 0.1% Triton X-100, rinsed, and saturated as above. Then they were incubated with primary antibodies diluted in blocking solution (PBS/BSA/serum) at 4°C overnight and with fluorescent secondary antibodies 30 min at room temperature. After immunoreactions all slides were rinsed twice in PBS for 15 min, mounted under a coverslip with Vectashield Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA), and photographed using Leica® fluorescence microscope. Antibodies, anti-human Keratin 14 rabbit polyclonal were from Covance, California, USA, anti-human Keratin 10 mouse monoclonal from Progen Biotechnik, Germany, and corresponding secondary antibodies (dilution 1/1,000) PE-conjugated anti-rabbit and FITC-conjugated anti-mouse were from Molecular Probes, Oregon, USA.
Western blot analysis
Western blot analysis was performed using ready-to-use NuPAGE® 4%–12% Bis–Tris acrylamide gels (Invitrogen, France) according to the supplier instructions. Blots were probed with appropriate primary antibodies and horseradish peroxidase-conjugated anti-secondary antibody (Bio-Rad, France). Immunodetection was performed using an ECL + chemiluminescence kit from Amersham.
Statistical analysis
All results are expressed as the mean ± SEM of at least 2 independent experiments performed in triplicate. Comparison between means was assessed using the Student’s t-test for unpaired data. If unequal variance was observed, the Welch’s correction was applied. A P value ≤0.05 was considered significant.
Results
Asymmetric distribution of EGFR in dividing skin keratinocytes
Given that normal keratinocytes expressing low levels of EGFR on the cell surface have stem cell-like properties [14] and that its asymmetric distribution during division of neural progenitors contributes to different cell fates [8], we used different approaches to assess EGFR distribution in dividing normal and cancer keratinocytes employing fluorescence microscopy. Images in Figure 1A show human keratinocytes displaying an uneven distribution of EGFR in the newly born daughter cells thereby illustrating asymmetric mitosis. Unequal allocation of EGFR was found in very few dividing cells in both normal (NHK) and cancer, squamous and basal cell carcinoma cultures (SCC and BCC, respectively). The observed asymmetric distribution of EGFR in normal and 2 different cancer keratinocytes corroborated, to some extent, the universality of the phenomenon in the epidermis and posed the question whether EGFR functions as a determinant of keratinocyte fate as it does in dividing neuronal cells [8].
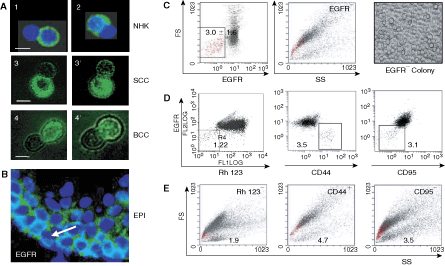
Keratinocyte cultures contain epidermal growth factor receptor (EGFR−) cells. (
EGFR− keratinocytes were rare and immature and excluded rhodamine 123
In normal human epidermis epidermal growth factor receptor was found mainly in the basal layer of proliferative keratinocytes and, to a lesser extent, in differentiating keratinocytes of the suprabasal layers (Fig. 1B). At closer inspection of the basal cell compartment, one could argue that there are single EGFR− cells. Assessment of the EGFR− versus EGFR+ populations in normal human epidermis (10 samples) by flow cytometry revealed that indeed EGFR− cells constituted a scarce proportion, that is 3.0% ± 1.6% of total live epidermal cells and 1.2% ± 0.6% of keratin 15 and/or 14 positive keratinocytes. They were of low forward scatter (FS) reflecting their small size and low side scatter (SS) reflecting their low granularity (Fig. 1C). We previously demonstrated that this size and structure profile identifies undifferentiated keratinocytes, which increase their size and granularity as they enter the differentiation program [20,21]. The light microscopy image, showing morphological uniformity and roundness of cells within a colony formed by primary EGFR− normal keratinocytes (Fig. 1C), confirmed undifferentiated state of EGFR− cells. Cells with this morphological profile were previously identified as side population (SP) of epidermal stem cells excluding Hoechst 33342 [22]. Interestingly, small EGFR− cells efficiently excluded Rhodamine 123 (Rh123), another SP marker shown to select for the most primitive hematopoietic stem cells and commonly used to determine a pool of quiescent stem cells [23 –26]. In addition, the small EGFR− cells described were positive for an epithelial stem cell marker, CD44 [27,28] but negative for proapoptotic marker CD95/Fas [29] (Fig. 1D). Backward FS and SS analysis of living Rh123−, CD44+ and CD95− (Fig. 1E) cells co-localized them with the EGFR− cells, hence showing that human normal epidermis contains a restricted population of small-sized EGFR−, Rh123−, CD44+, CD95− cells, with an immature undifferentiated phenotype.
An EGFR− subpopulation was found in skin tumors and in A431 SCC cell line
A rare EGFR− subpopulation (0.8%–3.7%) displaying an undifferentiated phenotype was also found in basal cell carcinoma (BCC) and SCC tumor samples as well as in the A431 SCC cell line. This intriguing finding prompted us to concentrate our efforts on defining EGFR− cells in the A431 cell line commonly used as a model of EGFR overexpressing cells, and compare the results with normal control keratinocytes. Cells with the EGFR− phenotype constituted 0.8% ± 0.4% of total live A431 cells (Fig. 2A) and were Rh123− in significant contrast to a majority of EGFR+ cells that were Rh123+ (Fig. 2B, P ≤ 0.001). However, as shown in Figure 2C, a small subpopulation of A431 EGFR+ cells also were Rh123−, indicating that the SP cell compartment was not homogeneous, and that EGFR may consent to a finer resolution within this compartment. Interestingly, the size of the EGFR+ Rh123− subcompartment was found significantly larger among the total live A431 cell population than in their normal counterparts (Fig. 2D, P ≤ 0.001), suggesting that one major difference between normal and A431 cancer cells may reside in this subcompartment.

Epidermal growth factor receptor (EGFR−) cells were quiescent in normal human keratinocytes (NHK) but actively cycling in A431 cell line cultures. (
EGFR− cells were more quiescent than their EGFR+ progeny in NHK but not in A431 cultures
Activation of the EGFR-signaling pathway stimulates cell exit from quiescence and its entry into the S-phase of the cell cycle [30,31]. Therefore, cells devoid of EGFR would be expected to remain quiescent. Flow cytometry analysis revealed that significantly more EGFR− than EGFR+ cells were in the G0/G1 phase of the cell cycle and significantly less in G2/M, demonstrating a greater cycling activity of the latter and an inclination mainly toward G0/G1 cell cycle arrest of the former, in both NHK (Fig. 2E) and A431 (Fig. 2F) cultures. Interestingly, however, a significantly higher number of EGFR− A431 cells than NHK was in the S-phase of the cell cycle (34.9% ± 6.4 % vs. 1.9% ± 1.0 % in NHK) likewise their cycling EGFR+ counterparts. This set NHK apart from A431 cultures, and suggested that EGFR− A431 cells enter the cycle with accelerated kinetics and that this process, apparently EGFR-independent, contributed to the generation of highly proliferating EGFR+ cells. Since a functionally important characteristic of adult stem cells is their quiescence [32 –34], the relative arrest of EGFR− cells at the G0/G1 phase of the cell cycle in conjunction with their immature morphology, SP phenotype, and their ability to divide asymmetrically as well as express the CD44 stem cell marker advocated for classifying them as stem-like cells. This assumption required confirmation by clonogenic and self-renewal assays.
Lack of EGFR on the cell surface correlated with the superior clonogenic and regenerative potentials of A431 cells
To determine the identity and functionality of rare EGFR− keratinocytes, we sorted the easily expandable A431 cells into EGFR− and EGFR+ fractions. Several parameters defining stem cells were then assessed in these fractions. Mackenzie et al. [27] argued that 3 criteria are sufficient to indicate the persistence of a stem cell pattern in vitro: generation and amplification of cell hierarchy, self-renewal, and differentiation. Intriguingly, despite the well-established connection between EGFR overexpression and uncontrolled proliferation of A431 cells [35,36], a clonogenic assay of sorted cells determined that it were the EGFR− not EGFR+ cells that formed colonies with a significantly higher efficiency (Fig. 3A, P ≤ 0.0001). Interestingly, the colonies formed by EGFR− cells were large, smooth-edged, and relatively homogeneous (Fig. 3B) resembling stem cell-derived holoclones described by Barrandon and Green [37]. An analysis of individual small and large A431 clones revealed that only large clones had the capacity to reform holoclones and give rise to small paraclones (Fig. 3B), thus reconstituting the heterogeneity of colonies formed by unsorted A431 cells (Fig. 3B). In contrast, paraclones produced only paraclones, known to be derived from transient amplifying, destined-to-differentiate progeny of stem cells [38]. Assuming that heterogeneity of colonies reflects the uneven proliferative potential of cells within the population [37], we concluded that the holoclone-forming EGFR− cells represent a stem-like cell compartment and further presupposed that a switch from the EGFR− to EGFR+ phenotype might be associated with a conversion of stem-like into fast amplifying cells contributing to an increase in cellular mass, to cellular hierarchy, and consequently, to the heterogeneity of A431 cultures.
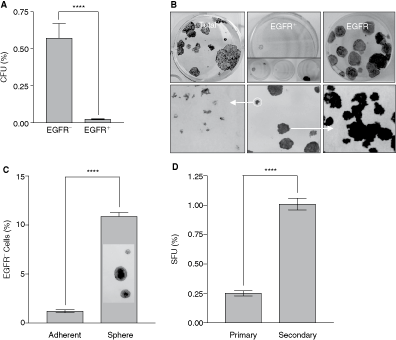
A431 sphere cultures favored expansion of the epidermal growth factor receptor (EGFR−) cells endowed with the superior to EGFR+ counterparts clonogenic and proliferative potentials (
Sphere-inducing conditions stimulate expansion of the EGFR− cell compartment
One attribute of stem cells is their ability to form spheres [39,40]. We questioned whether sphere growth conditions known to stimulate stem cell expansion could influence the size of the EGFR− compartment. Interestingly, as shown in Figure 3C, spheres contained a higher number of EGFR− cells than in monolayer A431 cultures (P ≤ 0.0001). Accordingly, when dissociated and replated under sphere-forming conditions more secondary spheres were formed (Fig. 3D), demonstrating that sphere conditions augmented the number of cells with the sphere-forming ability. The number of spheres was directly proportional to the size of the EGFR− cell compartment arguing further that EGFR− cells could constitute a pool of stem-like cells.
EGFR− cells self-renewed at a constant level and produced EGFR+ progeny
To address the relationship between EGFR phenotype and cell position in hierarchy, we reinforced our conclusion that an EGFR− subpopulation was enriched in stem-like cells by assessing self-renewal and differentiation abilities of EGFR− A431 cells. Colonies formed by sorted EGFR− cells were consecutively resorted and replated several times. As shown in Table 1, A431 EGFR− cells always generated EGFR+ cells dominating the regrowing cultures. Similar data obtained with 2 consecutive FACS fractionation of primary NHK, BCC, and SCC cell cultures generated in the lab from the respective tissue samples revealed that the ability of EGFR− cells to reproduce self and to generate numerous EGFR+ progeny is not restricted to the A431 cell line.
EGFR− C
FACS-sorted EGFR− cells were replated and cultured for 10 days before consecutive sorting. Data of 3 independent experiments of successive sorting plated in triplicate.
Interestingly, the percentage of EGFR− and EGFR+ cells was maintained repeatedly at a constant level (Table 1) suggesting that EGFR− cells divided asymmetrically to self-renew and to produce symmetrically dividing EGFR+ cells. The comparative genomic hybridization (CGH) analysis of DNA isolated from A431 EGFR− and EGFR+ cells (Supplementary Figs. 1 and 2; Supplementary materials are available online at http://www.liebertpub.com/) shows analogous chromosomal aberrations at high stringency, confirming their clonal origin and supporting the notion that EGFR+ cells were likely derived from EGFR− precursors. This conclusion was reinforced by experiments with sorted EGFR− GFP+ cells that acquired the EGFR+ GFP+ phenotype during culturing (Fig. 4A and 4B). Interestingly, EGFR+ cell-derived colonies always contained a small subpopulation (0.9%–1.2%) of EGFR− cells. Moreover, A431 cultures consisting of only EGFR+ cells, that is, depleted of EGFR− cells, reacquired the EGFR− component at levels similar to unsorted A431 populations (data not shown). Assuming a 100% purity of sorted cells, these data suggested that in the absence of the EGFR− component, a strictly defined portion of EGFR+ cells reverted to the EGFR− phenotype.
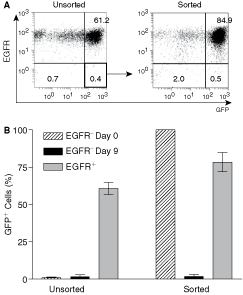
Epidermal growth factor receptor (EGFR−)/green fluorescent protein (GFP+) cells produced EGFR+/GFP+ progeny. (
Molecular differences between normal and cancer, and between EGFR− and EGFR+ cells: cell surface markers and gene expression profiles
Normal cells can provide insight into the behavior of cancer cells. To determine a possible functional relationship between EGFR abundance and cell fate, we compared redistribution of CD44, one of the most commonly used markers of normal and cancer stem cells [28,41], and CD95 in EGFR− and EGFR+ normal and A431 cancer keratinocytes. CD95 is expressed constitutively by basal keratinocytes [42], which are destined-to-differentiate progeny of epidermal stem cells. Hence in this study, CD95 served as a marker of committed-to-differentiate keratinocytes. Interestingly, flow cytometry analysis revealed that whereas in NHK the transition from EGFR− toward EGFR+ cells down-regulated CD44 and up-regulated CD95 (Fig. 1D), EGFR+ A431 cancer cells remained CD44+/low and CD95− (Fig. 5A). The observed correlation between the display of EGFR and both CD44 and CD95 expression is consistent with the previously established interaction between these factors [43 –45] and hinted that an exposure of CD44 and CD95 on the cell surface may be linked to a switch from EGFR− to EGFR+ phenotype, apparently malfunctioning in A431 cancer cells.
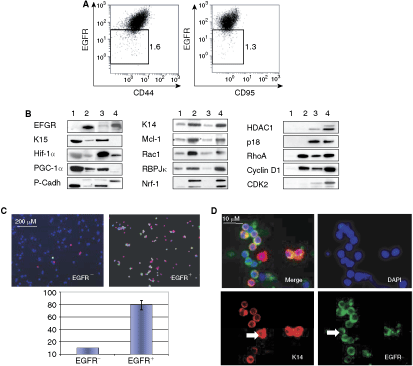
Epidermal growth factor receptor (EGFR−) and EGFR+ normal epidermal and A431 keratinocytes differed at the molecular levels. (
To define molecular events associated with the transition from the EGFR− to EGFR+ phenotype in cancer and normal keratinocytes, we subdivided A431 cells into EGFR− and EGFR+ fractions and performed a comparative study of their transcripts using SuperArray cDNA membranes profiling stem cell and apoptosis genes. The products of several differentially expressed transcripts (data not shown) were reanalyzed via western blotting (Fig. 5B). As expected, EGFR was not expressed in EGFR− NHK. It was, however, detectable in EGFR− A431 cells, though at a significantly lower level than in EGFR+ cells. This was consistent with our data in Figure 1A and suggested that similar to other cancer cells [46], A431 EGFR− cells may contain intracellular EGFR. Both normal and A431 cancer EGFR− cells expressed higher levels of keratin 15 (K15), a marker of epidermal stem cells [47,48], hypoxia-inducing factor 1α (Hif-1α), peroxisome proliferator-activated receptor γ co-activator-1 α (PGC-1α), and P-cadherin p55 form, than EGFR+ cells. Alternatively, the EGFR+ cells up-regulated K14, a marker of basal keratinocyte [49], anti-apoptotic Mcl-1, Rac1 belonging to Rho family of GTPases and implicated in keratinocyte differentiation [50], RBPJκ transcriptional repressor involved in the Notch-signaling pathway [51,52], and nuclear respiratory factor 1 (NRF-1) implicated in mitochondrial biogenesis [53]. Finally, we identified several proteins that were differentially expressed in A431 cancer and normal keratinocytes upon the switch from EGFR− to EGFR+ phenotype, including histone deacetylase 1 (HDAC1), p18INK4C, RhoA, cyclin D1, and CDK2, all of which are related to cell cycle regulation, suggesting that normal and cancer cells differ at the cell division level. Interestingly, some of the proteins (cyclin D1 and RhoA) not expressed by EGFR− NHK were up-regulated in EGFR− A431 cells, while others (p18INK4C) were uniquely expressed in the A431 cell line.
Because mouse epidermal stem cells located in the bulge of hair follicles already express K14 [54], we further assessed K14 expression in EGFR− and EGFR+ subpopulations of human normal (Fig. 5C) and A431 (Fig. 5D) keratinocytes by immunohistochemistry. As expected from a marker of the basal cell compartment, K14 was mainly expressed by keratinocytes expressing high levels of EGFR. Still, keratinocytes expressing both markers in any combination could be found, confirming that K14 is not an exclusive marker of stem cells. However, the presence of EGFR− K14+ A431 cells is coherent with the presumed stemness of keratinocytes devoid of surface EGFR.
To summarize, we demonstrated that normal epidermis and cancer keratinocyte cultures, of mainly A431 SCC but also primary SCC and BCC cells, contain a small subpopulation of EGFR− cells endowed with stem-like properties, including high clonogenic and proliferative potential, expression of stem cell markers, propensity for quiescence, and competence to produce phenotypically and functionally distinct daughters. That EGFR− cells may function as stem-like cells was further supported by our supplementary data (Supplementary Fig. 3) showing that EGFR− but not EGFR+ BCC cells were capable of reconstructing multilayered, although morphologically abnormal, epidermis in 3D skin equivalent cultures known to recapitulate the in vivo-like program of keratinocyte differentiation.
Discussion
The findings of this study convey 2 major messages: first that EGFR can be asymmetrically distributed in dividing keratinocytes and prospectively may function as an epithelial cell fate determinant, and second that a small subpopulation of cancer keratinocytes endowed with stem cell attributes may be the keratinocytes devoid of surface EGFR. The deduced interpretation of our finding led us to propose a model in which an EGFR− cell would retain quiescence and divide infrequently both to reproduce self (to maintain a constant G0 pool) and an actively cycling EGFR+, hence responsive to EGFR mitogenic signals, daughter cell (to replenish the keratinocyte population). In our proposed model, the acquisition of EGFR on the cell surface would locate a critical point of the phenotypic and functional switch between these 2 fates.
This model is strongly supported by our data and is consistent with the general understanding of the vital role of EGFR in development, proliferation, differentiation, and survival of keratinocytes evidenced by the severity of phenotypes in mice with compromised EGFR expression [11 –13,55 –58]. The epidermis in these cases was immature and thin with fewer layers of abnormally differentiating cells and with an increased apoptotic index. If EGFR− cells were precursors of EGFR+ cells as we suggest, the compromised EGFR function would not affect the EGFR− reproductive compartment but would target the compartment of EGFR+ progeny leading to the exact same findings as in the EGFR-deficient mouse models and in anti-EGFR-treated cancer patients [11,15,55 –58].
In the epidermis, EGFR-displaying cells are primarily destined-to-differentiate transient amplifying daughters of stem cells located in the basal layer [11 –15,56]. After a short period of intense proliferation, the EGFR+ cells, while moving to the suprabasal layer, enter the differentiation program [11 –13] linked to the cell death pathway [20,21]. Conceivably, to stay quiescent and undifferentiated, a stem cell should inactivate the EGFR-signaling pathway. We provide evidence demonstrating that in the epidermis and in A431 SCC cell line this may be achieved, though not exclusively, by evading EGFR on the cell surface. Accordingly, in our preliminary xenograft experiments, only EGFR− cells were tumorigenic in SCID mice (data not shown).
A role of EGFR in directing the fate of keratinocytes was first indicated by studies in which 20% of epidermal keratinocytes with the lowest content of surface EGFR displayed traits of epidermal stem cells [14]. Our study not only confirmed these findings but also refined stemness to 0.8%–1.2% of EGFR− of normal and A431 cancer keratinocytes. This frequency approximates the occurrence of long-term repopulating cells found in human epidermis by Schneider et al. [59]. Whether a subpopulation of EGFR− Rh123− keratinocytes represents the long-term repopulating cells remains to be experimentally validated. Interestingly, however, consistent with the consensus that the repopulating potential of stem cells is directly linked to their cycling quiescence [32,60,61], the majority of EGFR− keratinocytes was confined to G0/G1 cell cycle arrest and was residing within the Rhodamine 123 excluding subpopulation commonly associated with the SP compartment of quiescent stem cells [23 –26]. This suggests that the EGFR− Rh123− phenotype may define the quiescent subpopulation of stem keratinocytes.
The ability of the EGFR+ cell-derived cultures to reestablish the EGFR− cell compartment is particularly interesting. We suggest that in our experimental settings some of the EGFR+ cells may be capable of reverting to the EGFR− phenotype to compensate for loss of the EGFR− cell compartment. One mechanism that can cause such cell reprogramming involves resetting quiescence of cycling stem cells [60 –63] reviewed recently by Ratajczak [61]. Wilson et al. [60] showed that hematopoietic stem cells (HSC) can oscillate between 2 functional states, a quiescent CD34− state and an active CD34+ state. Ratajczak [61] also pointed that CD34 expression reflects the activation/kinetic state of HSC. The main point stressed by both is that the shift from quiescent to activated HSC is reversible. This reversibility of HSC appears to be their inherent property and Glauche et al. [63] propose a mathematical model in which stem cells reversibly adopt their proliferation/quiescent state to systemic needs [63]. In our model, we speculated that A431 EGFR− quiescent stem cells may acquire EGFR on the cell surface as they become activated into the cell cycle and propose that the EGFR+ cell population, consisting mainly of transit amplifying cells, also includes a small proportion of activated, fast dividing stem cells able to return to the quiescent state. A depletion of EGFR− stem-like cells may be sensed at the EGFR+ population level as a disruption of homeostasis triggering reversion of EGFR+ fast dividing stem cells into a quiescent, critical for preserving self-renewal, EGFR− state. The reversibility of stem cell status could explain why starting from either EGFR− or EGFR+ sorted cells, the same steady-state ratio of EGFR− and EGFR+ subpopulations is obtained after 9 days of culturing. The constant value of the final ratio, identical to that in unsorted cultures, strongly suggests that we are not dealing with an accidental product of culture conditions but with some inherent properties of the cell population essential for its perpetual renewal. In this respect, the ability of sorted EGFR+ cells to reestablish the EGFR− cell compartment resembles replenishment of empty stem cell niches in vivo [64 –66]. The presence of EGFR+ cells within the Rh123− subpopulation favors such interpretation. Minute in number in NHK but overrepresented in the A431 cancer cell line, the Rh123− EGFR+ component mirrored the frequency of EGFR− cells in S-phase, implying a mechanistic link between the activation into cell cycle and EGFR acquisition. Such a link is supported by the observed asymmetric distribution of EGFR at mitosis and by the apparent lack of EGFR− cells in the G2/M phase, placing the EGFR acquisition after S-phase.
The switch from EGFR− to EGFR+ was accompanied by the expression of genes involved in DNA replication and cell cycle progression. Interestingly, however, and consistent with differences in the EGFR− cell cycle kinetics, the expression pattern of these genes differed between normal and A431 EGFR− cells, pointing to the possible signaling pathways that may be involved in the reversibility of the quiescent/activated state of these cells. Particularly interesting is the striking difference in RhoA expression, a coordinator of cytoskeleton organization during cell division [67], and in cyclin D1, a downstream effector of RhoA, suggesting a reverse relationship between their presence and cell quiescence. Also, high levels of Hif-1α and its upstream inducer PGC-1α [68] in EGFR− reproductive cells appears interesting in this context. Both acting in concert to suppress mitochondrial function [69 –71], a prerequisite for stem cell quiescence [69,71,72], may be responsible for relative quiescence of stem keratinocytes. In favor of this suggestion is the recently discovered c-Myc/Hif-1α crosstalk contributing to cell cycle arrest [73,74] and data showing that mitochondria are less developed in adult stem cells than in their differentiating progeny [75]. Indirect support for the possible involvement of Hif-1α in imposing the A431 cell quiescence also provides the observation that hypoxia-prone sphere cultures stimulated expansion of the EGFR− cell compartment.
The ability of epidermal stem cells to oscillate between quiescence and proliferation has been recently documented in mouse skin where NFATc1 transcription factor negatively regulating CDK4 was identified as the key factor reinforcing quiescence, thereby protecting stem cells from exhaustion [32]. We speculate that, analogous to NFATc1, shunning of surface EGFR preserves EGFR− cells quiescence and consequently their long-term maintenance, simply by rendering them unresponsive to mitogenic stimuli. While this seems to apply to NHK, the accelerated cell cycle kinetics of A431 EGFR− cells setting apart cancer and NHK points to the existence of EGFR-independent mechanism(s) operating in A431 cells that allows EGFR− stem-like cells to overcome quiescence without exhaustion. Although these protective mechanisms remain to be elucidated, an intriguing presence of p18INK4c in cancer A431 but not in normal keratinocytes suggests that this cell cycle inhibitor may guard asymmetric divisions of cycling A431 cells and block their differentiation as it does in neuronal cells [76]. Intrinsic and extrinsic factors involved in the control of quiescence/activation have just started to be identified in hematopoietic and epidermal stem cells [32,77 –79]. Obviously, niche that provides a specific signaling context is playing a key role in the control of stem cells activity [66], oscillation between dormant and proliferative states, and transition to differentiation under normal conditions [78], and as accumulating evidence suggest, niches also influence the function of cancer stem cells, tumor invasiveness, and metastasis [80].
Our discovery that the A431 cell line contains a small subpopulation of stem-like EGFR− cells is by no means surprising, since the persistence of cancer stem cells has been documented for most human cancer cell lines established from tumors of different tissues [27,41,81 –83]. Cancer cell lines, despite many drawbacks, still remain a valuable experimental model to study cancer stem cells [for discussion, see Ref. 84].
In conclusion, one possible mechanism controlling diversity between long-term multipotent stem cells and their progenitors would be a selective avoidance of growth factor receptors on the cell surface since they do not respond to the same growth factors [85]. From our present work we argue that keratinocyte fate determination may be regulated at the level of asymmetric mitosis by uneven distribution of surface EGFR between 2 daughter cells and provide evidence that this unequal EGFR segregation contributed to their molecular and functional divergence. Asymmetric EGFR appears to balance the stem cell quiescence and proliferation/differentiation that clearly malfunction in cancer cells. Furthermore, we distinguished 3 cell populations in keratinocyte cultures: a small but constant pool of quiescent Rh123− EGFR− cells, that did not differ dramatically between normal and cancer cultures, a small but variable in size between cancer and normal cultures pool of Rh123− EGFR+ cells representing perhaps activated stem cells and finally, a major pool of Rh123+ EGFR+ transient amplifying cells. Whether these 3 different pools represent a hierarchy of reproductive potential of long- to- short-term epidermal stem cells and their differentiating progeny remains to be discovered. We believe that our findings identified a novel role of EGFR in keratinocyte fate determination and may provide mechanistic insights into carcinogenesis and the development of resistance to current anti-EGFR therapies.
Footnotes
Acknowledgments
We are thankful to Martine Tual for her skilful technical assistance, to Marie Estienney for skin cancer cultures, and to Harold Fauvel for his valuable help with video microscopy. This research was sponsored by Institut National de la Santé et de la Recherche Médicale INSERM, Conseil Régional Nord Pas de Calais, 3M Health Care, Groupement des Entreprises Françaises dans la Lutte contre le Cancer GEFLUC, Institut de Recherche sur le Cancer de Lille (IRCL), Société Française de Dermatologie (SFD), and Institut national du Cancer (INCa). H.L. is a recipient of a grant from region Nord Pas de Calais/INSERM and from IRCL. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.