
Abstract
Mesenchymal stem cells (MSCs) with their multilineage developmental plasticity comprise a promising tool for regenerative cell-based therapy. Despite important biological properties, which the MSCs from different sources share, the differences between them are poorly understood. Hence, it is required to assign a molecular signature to each of these MSC populations, based on stem cell related genes and early lineage or developmental markers. Understanding their propensity to differentiate to different lineages is fundamental for the development of successful cell-based therapies. Culture expansion of MSCs is a prerequisite, since high absolute numbers of stem cells are required to attain a clinical dose. Here, we compared the different culture conditions for long-term expansion of human MSCs isolated from the Wharton’s jelly (WJ) of the umbilical cord while preserving their stem cell characteristics and differentiation potential. We find that DMEM-KO and DMEM-F12 are superior as compared to the other media tested in supporting the in vitro expansion of the WJ-MSCs. We studied the gene expression profile of WJ and bone marrow-derived MSCs (BM-MSCs) both at early and late passages using Human Stem Cell Pluripotency Array, and our data revealed differences at the transcriptional level between the two MSC types. Compared to BM-MSCs, WJ-MSCs had higher expression of undifferentiated human embryonic stem cell (hES) markers like NANOG, DNMT3B, and GABRB3, pluripotent/stem cell markers, as well as some early endodermal markers both at early and late passages. To conclude, WJ-MSCs possess properties of true stem cells, which they retain even after extended in vitro culturing.
Introduction
A
MSCs have been isolated conventionally from bone marrow [1]. However, there is enough good evidence that MSCs exist not only in bone marrow, but also virtually in all the organs of body. These include adipose tissue [7], periosteum [8], tendon [9], dental pulp [10], synovial membrane [11], skin [12], amniotic fluid [13], umbilical cord [14], umbilical cord blood [15], limbal tissue [16], and menstrual blood [17]. Tissue source of the MSC may be critically important in determining biological activity. Different tissue source may generate MSC products with different cytokine expression profile [18]. Hence, different MSC tissue source may be especially suited for specific clinical applications.
Therefore, in recent years, there has been a lot of interest in the search for novel and potent stem cell sources in the human body that may be suited for specific clinical applications, and the umbilical cord is one such source. The umbilical cord connects the placenta and the fetus during pregnancy and is usually discarded after delivery. It can provide an inexhaustible source of stem cells for therapy without involving any invasive procedures or ethical concerns. MSCs have been isolated from different compartments of the umbilical cord, and Wharton’s jelly (WJ) is the embryonic mucous connective tissue lying between the amniotic epithelium and the umbilical vessels. The umbilical cord is a fetus-derived tissue and the WJ-derived MSCs share some properties unique to fetal derived MSCs, like having faster proliferation and greater ex vivo expansion capabilities than adult MSCs [19,20].
WJ-MSCs are therapeutic in several different preclinical animal models of human disease such as neurodegenerative disease, cancer, diabetes, heart disease, etc [21 –24]. To take this initiative to the next step, first, it is important to generate a clinical quantity of MSCs, while retaining stem cell characteristics, immunosuppressive capacities, and multilineage differentiation potential. Reaching the estimated clinical dose of 2–5 × 106 cells per kilogram body weight of adult patients can not only be challenging but could also hamper the progress of clinical WJ-derived MSC applications. Hence, in vitro expansion of MSCs is a prerequisite for MSC transplantation. Not much has been reported about therapeutic expansion of WJ-derived MSCs. Media used for tissue culture would have an important impact on growth and differentiation of MSCs.
Second, though MSCs from different origins share important biological properties, it is not clear which type of adult stem cell or MSC should be selected for the different therapeutic approaches. The differences between MSCs from various sources are poorly understood. The origin or source of MSCs may determine their fate and functional characteristics. The important question being whether MSCs from different origins exhibit unique gene expression profiles, which could explain their differentiation characteristics. Analyzing mRNA expression levels is a powerful approach that can be used to understand the propensity and capacity of MSCs from a particular source to differentiate toward a particular lineage or adopt a certain fate.
In this present study, we attempt to identify the optimal basal culture media for long-term expansion of MSCs derived from WJ, which could lead to the production of large numbers of MSCs for subsequent cellular therapeutic approaches. We have performed growth kinetics studies, analyzed surface marker phenotype, and studied differentiation potential till late passages in the different basal media. At late passages, we checked these cells for senescence, transformation markers, and looked for chromosomal abnormalities by karyotyping. We tried to upscale these WJ-MSCs to a clinical dose. Finally, we have identified a distinctive gene expression profile for the WJ-MSCs, with BM-MSCs as control, with respect to stem cell and early lineage markers with an aim to understand better their self-renewal capacity and propensity to differentiate toward different lineages or cell types in vitro or in vivo. Also, we have examined if the gene expression levels of these markers remain stable during continuous culturing in vitro for these two MSC populations.
Materials and Methods
Isolation and culture of WJ-MSCs
Human umbilical cords (n = 15) from both sexes were collected from full-term births after either cesarean section or normal vaginal delivery with informed consent using the guidelines approved by the Institutional Committee for Stem Cell Research and Therapy (ICSCRT) and Institutional Ethics Committee (IEC) at the Manipal Hospital, Bangalore, India. MSCs from WJ of umbilical cord were isolated as previously described [24]. Briefly, the cord was cut into 2–3 cm segments. Following disinfection with 70% ethanol for 30 s, the umbilical cord vessels were stripped manually from cord segments and the exposed mesenchymal tissue was treated with collagenase blend type H (Sigma-Aldrich, St. Louis, MO) at 37°C for 16 h followed by 0.05% trypsin (Invitrogen Carlsbad, CA) at 37°C for 10 min. Next, the cells were suspended in Knock out Dulbecco’s modified Eagle’s medium (DMEM-KO) (Invitrogen) and 10% FBS (Hyclone) and plated on tissue culture plastic plates. All cultures were incubated at 37°C with 5% humidified CO2 and passaged when they reached ∼70% confluence. For experiments utilizing early passage WJ-MSCs, cultures at passage 3–5 were used; while for late passage, WJ-MSCs cultures at passage 15–20 were used.
WJ-MSCs were cultured in the following media: DMEM with 1,000 mg/mL glucose (DMEM-LG) and 2 mM
Human bone marrow samples were taken after written consent, using guidelines approved by the IEC at the Manipal Hospital, Bangalore, India. Bone-marrow (BM) MSC cultures were established from four donors as previously described [25]. The mean age of BM donors was 27.5 years (range: 20–35). For PCR array experiments utilizing early passage BM-MSCs, cultures at passage 3–5 were used while for late passage BM-MSCs cultures at passage 15–18 were used.
MSCs used for characterization, upscaling, and array experiments, at early as well as late passages, were grown under identical culture conditions, including plating density, tissue culture plastic, confluency, etc.
Growth kinetics
Growth kinetics was analyzed by calculating population doubling and population doubling time. Population doublings were calculated using the formula: X = [log10(NH)- log10(NI)]/log10(2) [26], where NI is the inoculum cell number and NH the cell harvest number. The population doubling time was obtained by the formula: TD = tplg2/(lgNH-lgNI). NI: the inoculum cell number; NH is the cell harvest number, and t is the time of the culture (in hours).
Senescence assay
Senescence assay was performed with WJ-MSC cell cultures using Senescence β-Galactosidase Staining kit (Cell Signaling, Danvers, MA), according to the manufacturer’s protocol. Briefly, cells were grown in 35 mm plate, washed with PBS, fixed, and stained using 1 mL staining solution in citrate buffer (pH 6.0) overnight at 37°C. Cells were observed for development of blue color under a microscope using 10/20× objective. Five random fields each from four biological replicates of WJ-MSCs were used to calculate the percentage of positive cells.
Immunophenotyping
WJ-MSCs cultured in the different basal media were taken for flow cytometric analysis at early and late passages. The following antibodies were used to mark the cell surface epitopes—CD90-PE, CD44-PE, CD73-PE, CD166-PE, CD34-PE (all from BD Pharmingen, San Diego, CA), and CD105-PE (R&D Systems Inc., Minneapolis, MN). Cells from 5-cell stack (Corning, Corning, NY) experiment were stained with additional antibodies CD45-FITC (BD Pharmingen) and HLA-DR-FITC (BD Biosciences). All analyses were standardized against negative control cells incubated with isotype-specific IgG1-PE and IgG1-FITC (BD Pharmingen). The number of cells staining positive for a marker was determined by the percentage of cells present within a gate established such that 5% of the positive events measured represented nonspecific binding by the PE or FITC-conjugated isotype-matched control. At least 10,000 events were acquired on BD LSR II flow cytometer and the results were analyzed using WIN MDI v2.8 software.
Differentiation
Osteogenic differentiation was induced in the different basal media supplemented with 10% (vol/vol) FBS (Hyclone), 0.1 µM dexamethasone, 10 mM β glycerophosphate, 0.2 mM ascorbic acid (all reagents from Sigma Aldrich). Mineralized deposits were visualized by Von Kossa staining after 21 days. Cells were fixed with 4% Paraformaldehyde (Sigma Aldrich) for 20 min and exposed to 1% silver nitrate (Merck, Gibbstown, NJ) under bright light for 60 min. For adipogenesis, cells were induced for differentiation in the different basal media supplemented with 10% (vol/vol) FBS, 200 µM indomethacin, 0.5 mM 3-Isobutyl -1-methylxanthine, 10 µg/mL insulin, and 1 µM dexamethasone (all reagents from Sigma Aldrich). At day 18, the presence of lipid droplets was confirmed by Oil Red O stain (Sigma Aldrich). Images were captured using Nikon Eclipse TE2000-U inverted microscope (Nikon, Tokyo, Japan). To quantitatively analyze osteogenic differentiation, five random fields in three biological replicates of WJ-MSCs for each medium condition were counted for mineral deposits by using Image-Pro AMS version 6.0 software (Media Cybernetics, Inc, Silver Spring, MD).
Reverse transcription-polymerase chain reaction analysis (RT-PCR)
Total cellular RNA was isolated using an RNeasy mini kit (Qiagen, Hilden, Germany). To eliminate traces of genomic DNA, the RNA samples were treated with DNA-free DNase I (Ambion, Austin, TX), according to manufacturer instructions. The RNA was reverse-transcribed into cDNA using high capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) or Superscript III reverse transcriptase (Invitrogen), according to the manufacturer’s instructions. PCR amplification was performed using Taq Polymerase (Sigma) by following standard procedures. The primer sequences were as follows: c-Myc: forward primer, 5′-AAGACTCCAGCGCCTTCTCTC-3′ and reverse primer, 5′-GTTTTCCAACTCCGGGATCTG-3′, (526 base pairs [bp]) ERCC3: forward primer, 5′-CCAGGAAG CGGCACT ATGAGG-3′ and reverse primer, 5′-GGTCGTCC TTCAGCGGCATTT-3′, (171 bp); RAD51: forward primer, 5′-TTTGGAGAATTCCGAACTGG-3′ and reverse primer, 5′-AGGAAGACAGGGAGAGTCG-3′, (588 bp); XRCC4: forward primer, 5′-AAGATGTCTCATTCAGACTTG-3′ and reverse primer, 5′-CCGCTTATAAAGATCAGTCTC-3′,(233 bp); P16: forward primer, 5′-TTATTTGAGCTTTGGTTCTG-3′ and reverse primer, 5′-CCGGCTTTCGTAGTTTTCAT-3′, (354 bp); P21: forward primer, 5′-GAGGCCGGGATGAGTTG GGAGGAG-3′ and reverse primer, 5′-CAGCCGG CGTTTGGAGTGGTAGAA-3′, (220 bp); P53: forward primer, 5′-TTGGATCCATGTTTTGCCAACTGGCC-3′ and reverse primer, 5′-TTGAATTCAGGCTCCCCT TTCTTGCG-3′, (488 bp); 18s: forward primer, 5′-CGGCTA CCACATCCAAGGAA-3′ and reverse primer, 5′-GCTGG AATTACCGCGGCT-3′, (186 bp).
Quantitative PCR
The amplification reaction was performed using Taqman Universal Master Mix and Assay-On-Demand Taqman primer/probe sets (Applied Biosystems), according to manufacturer’s protocol in the ABI 7500HT RT-PCR system (Applied Biosystems). The assays for Oct4 and ABCG2 were Assay IDs Hs00742896_s1 and Hs00184979_m1, respectively. Eukaryotic 18S rRNA (Assay ID Hs99999901_s1) was used as an internal control in all assays. SDS v2.1 software was used to analyze the results. All PCRs were performed in duplicates. The results are presented as means of the duplicate measurements.
Immunofluorescence staining
WJ-MSCs were grown on glass coverslips or chamberslides (BD Falcon, San Jose, CA). They were fixed with 4% paraformaldehyde (Sigma) for 20 min at room temperature. They were permeabilized in 0.3% Triton X-100 and blocked in 10% normal rabbit serum (Sera Laboratories International Ltd, West Sussex, UK). The primary and secondary antibody reactions were carried out using standard protocols. The primary antibodies used were goat anti-Oct4 (1.6 µg/mL, R&D Systems), mouse anti-ABCG2 (1:40; Sigma) and mouse anti-Vimentin (1:100; BD Biosciences Pharmingen). Secondary antibodies used were FITC-conjugated rabbit anti-goat IgG (1:250; Chemicon, Temecula, CA) and Cy3 conjugated rabbit anti-mouse IgG (1:250; Chemicon). Slides were counterstained with 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) for 5 min. Fluorescent images were captured using a Nikon-Eclipse-90i microscope (Nikon) and Image-Pro AMS version 6.0 software.
Karyotype analysis
Karyotyping of WJ-MSCs cultured in different basal media was performed at early and late passages. Standard Giemsa staining procedure was carried out. At least 20 metaphase spreads were analyzed from each MSC preparation. Images were acquired using Nikon-Eclipse-90i microscope (Nikon).
Taqman low density arrays
For analyzing the expression of a focused panel of pluripotent and stem cell markers, the Human Stem Cell Pluripotency Array (Applied Biosystems, Foster City, CA) containing a well-defined set of validated gene expression markers to characterize embryonic stem (ES) cell identity was used. The 384 wells of each Human stem cell pluripotency array card were preloaded with fluorogenic probes and primers (Applied Biosystems). cDNAs were loaded on the microfluidic cards for thermal cycling on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). Expression values for target genes were normalized to the expression of 18s rRNA. Transcriptional analysis was performed on early and late cultures of WJ-MSCs and BM-MSCs. For data analysis, the ABI PRISM 7900HT Sequence Detection System software (SDS v2.2 software) calculated the levels of target (WJ-MSC) gene expression in samples relative to the level of expression in the calibrator (BM-MSC) with the Comparative Ct Method (ΔΔCt). A cut off cycle threshold value of 35.0 was arbitrarily assigned. Samples with a cycle threshold of 35 or less were considered for calculating the fold change in expression.
Quantitative RT-PCR confirmation of array data
To confirm the gene expression profile obtained by TLDA, a number of select genes were subjected to quantitative and semiquantitative RT-PCR analysis, using pooled cDNAs derived from the four different samples used for the PCR array experiment. Quantitative amplifications were carried out in duplicate using SYBR green master mix (Applied Biosystems). PCRs were run on an ABI Prism 7500HT (Applied Biosystems), and SDS v2.1 software was used to analyze the results. All measurements were normalized by 18s rRNA. The sense and antisense primers used for each gene were as follows: Desmin: forward primer, 5′-CCAACA AGAACAACGACG-3′ and reverse primer, 5′-TGGTATG GACCTCAGAACC-3′, (407 base pairs [bp]); GATA4: forward primer, 5′-TCCAAACCAGAAAACGGAAG-3′ and reverse primer, 5′-CTGTGCCCGTAGTGAGATGA-3′, (186 bp); hGABRB3: forward primer, 5′-CCTTGCCCAAAATCCCCTATG TCAAAGC-3′ and reverse primer, 5’-GTATCGCC AATGCCGCCTGAGACCTC-3′, (276 bp); Nestin: forward primer, 5′-CAGCGTTGGAACAGAGGTTGG-3′ and reverse primer, 5′-TGGCACAGGTGTCTCAAGGGTAG-3′, (388 bp); REX1: forward primer, 5′-TGAAAGCCCACATC CTAACG-3′ and reverse primer, 5′-CAAGCTATCCTCCTGC TTTGG-3′, (553 bp); SOX17: forward primer, 5′-ACATGAA GGTGAAG GGCGAG-3′ and reverse primer, 5′-TTGTAGTT GGGGTGGTCCTGC-3′, (285 bp).
Statistical analysis
Data were presented as means ± standard deviation. Statistical comparisons were performed using student’s t-test. P values <0.05 were considered significant.
Results
To determine the optimal basal medium for expansion and long-term culture of WJ-MSCs, MSCs from human umbilical cords were isolated and allowed to attach in DMEM-KO medium, since this was reported to be the optimal medium for isolation and culture of BM-MSCs by our group previously [25]. Once they attained enough cell numbers, that is, at passage 1–2 (since at passage 0 we get only ∼5–6 × 104 cells/cord), they were plated at 5,000 cells/cm2 in 35 mm dish in the presence of different basal media as described under Materials and Methods. Justification of the four basal media selected for the expansion of WJ-MSCs being, DMEM-HG and DMEM-F12 are the most commonly used basal media for the culture of MSCs. To assess the effect of low glucose concentration on the proliferation of WJ-MSCs, DMEM-LG was tested. As embryonic stem (ES) cells are most often cultured in DMEM-KO, we decided to test this for the WJ-MSCs.
Selection of optimal basal medium for expansion and growth characteristics of WJ-MSCs
To assess the proliferation capacities of the WJ-MSCs cultured under different media conditions, growth curves were plotted. For this, the total number of cells at each passage was calculated as a ratio of total number of cells harvested to total number of cells seeded multiplied by the total number of cells from the previous passage. This led us to calculate the total numbers of cells we would have harvested if we had cultured all cells from each passage. The long-term growth curves varied considerably between the different WJ samples for each of the media tested, which resulted in maximal cell numbers of 8.6 × 1011 to 8.6 × 1015 for DMEM-KO, 5.4 × 1012 to 1.9 × 1016 for DMEM-F12, 1.4 × 109 to 4 × 1014 for DMEM-HG, and 7.5 × 106 to 6 × 108 for DMEM-LG (Fig. 1B).
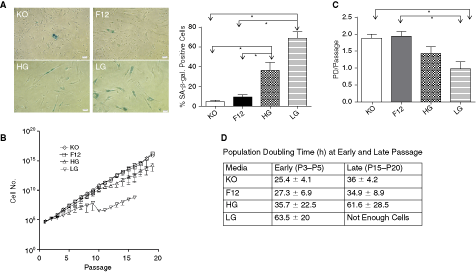
Growth characteristics and senescence in WJ-MSCs. (
Greater growth potential of WJ-MSCs cultured in DMEM-KO and DMEM-F12 was also evident from the number of population doublings, which were determined at each subcultivation, and mean values were plotted. WJ-MSCs cultured in DMEM-KO, DMEM-F12, DMEM-HG, and DMEM-LG corresponded to a mean of 1.89 ± 0.28, 1.95 ± 0.3, 1.4 ± 0.46, and 0.99 ± 0.44 population doublings, respectively (Fig. 1C). Cultures in DMEM-KO and DMEM-F12 had significantly higher proliferative potential than those in DMEM-LG (P < 0.01).
The mean population doubling time of early (P3–P5) and late (P15–P20) cultures of WJ-MSCs grown in DMEM-KO, DMEM-F12, DMEM-HG, and DMEM-LG was calculated and is shown in Figure 1D. WJ-MSCs cultured in DMEM-HG (35.7 ± 22.5 h) and DMEM-LG (63.5 ± 20 h) had a longer mean doubling time as compared to those cultured in DMEM-KO (25.4 ± 4.1 h) and DMEM-F12 (27.3 ± 6.9 h). Mean doubling time for early cultures in DMEM-KO and DMEM-F12 differed significantly than those cultured in DMEM-LG (P < 0.01). No significant changes were observed between early and late passages with regard to the time required for population doubling for each of the four media tested.
Again, the WJ-MSCs cultured in DMEM-KO and DMEM-F12 were small in size and maintained the typical spindle-shaped fibroblast-like morphology till late passages, whereas those cultured in DMEM-HG and DMEM-LG appeared flat, broad, and granular from the 5th passage onward.
During in vitro culture MSC possess a limited life span and finally undergo replicative senescence, which is characterized by cell cycle arrest, telomere shortening, and altered morphology. To look for replicative senescence, we have employed the enzyme lysosomal pH6 β-galactosidase (SA-β-gal) as a senescence marker. The percentage of SA-β-gal positive cells and the intensity of staining were higher in MSCs cultured in DMEM-HG (36.1% ± 20.3%) and DMEM-LG (69.1% ± 19.3%) media as compared to those cultured in DMEM-F12 (9.4% ± 6.9%) and DMEM-KO (4.9% ± 3.8%), both at early (data not shown) as well as late passages (Fig. 1A). There was a significant difference in the percentage of positive cells (P < 0.05) between DMEM-KO/F12 and DMEM-HG or DMEM-LG.
Immunophenotypic characterization of WJ-MSCs
WJ-MSCs from 3 to 5 different umbilical cord samples at early and late passages were characterized by flow cytometry. A panel of six markers was tested. All WJ-MSC preparations cultured in DMEM-KO, DMEM-F12, DMEM-HG, or DMEM-LG were negative for hematopoietic marker CD34 and were consistently positive for CD44, CD73, CD90, CD105, and CD166, which are known to be expressed on MSCs (Table 1). Percent expression of these markers was quite similar in all the four media at early passage; while at late or beyond 15 passages, the detection level for CD166 was lower as compared to early passage though a complete shift was still observed. MSCs cultured in DMEM-LG could not be analyzed for these markers at late passages as enough cell numbers could not be generated for flow cytometric analysis.
The table shows mean values of %-positive cells ± standard deviation to the total number of cells analyzed (n ≥ 3).
Abbreviation: WJ-MSC, Wharton’s jelly-derived mesenchymal stem cell.
In vitro multipotential differentiation of WJ-MSCs
To investigate the multilineage differentiation potential of WJ-MSCs at early and late passages when cultured under different media conditions, we directed the cells toward adipogenic and osteogenic lineages. Adipogenic differentiation was identified by Oil red O staining while osteogenic differentiation was detected by Von Kossa staining. When the WJ-MSCs were cultured in the four different basal media, DMEM-KO, DMEM-F12, DMEM-HG, and DMEM-LG, they retained their capacity to differentiate to adipocytes and osteoblasts till late passages (Fig. 2). Adipogenic differentiation at late passage was comparable in all the four different basal media tested. Osteogenic differentiation took place for WJ-MSCs cultured in all the four basal media at late passage but calcification was the highest in DMEM-KO (Fig. 2E–2H). In an attempt to quantitate osteogenic differentiation, the percentage of mineral deposit area was calculated at late passages and were 12.4% ± 11.5% for DMEM-KO, 3.6% ± 2.2% for DMEM-F12, 8.5% ± 3.1% for DMEM-HG, and 7.1% ± 4.6% for DMEM-LG. The mineralization in DMEM-KO was significantly higher (P < 0.05) than in DMEM-F12. For all cultures, induced cells showed significantly stronger staining than noninduced control cells (Fig. 2).
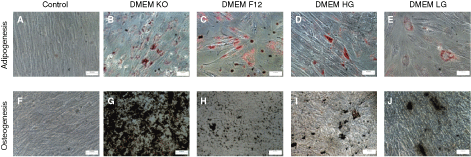
Confirmation of multilineage differentiation potential of WJ-MSC till late passages. The late passage (P15–P20) cells were investigated for in vitro multilineage differentiation capacity in the different media. Adipogenesis was detected by neutral oil droplet formation stained with Oil Red O at day 18 (
Detection of pluripotent/stem cell markers at mRNA and protein level
To study the expression of pluripotent stem cell markers, we evaluated the WJ-MSCs cultured under different media conditions and at early and late passages for the expression of ABCG2 and OCT-4 at the mRNA level by quantitative RT-PCR and at the protein level by immunofluorescence (Fig. 3). Oct-4 is a marker for pluripotency while ABCG2 belongs to the ATP-binding cassette superfamily of transmembrane proteins and is ubiquitously expressed in various stem cells (hematopoietic, muscle, neural, and testicular) [27]. As shown in Figure 3, WJ-MSCs expressed pluripotency/stem cell markers OCT-4 and ABCG2. The gene expression levels for OCT-4 and ABCG2 did not statistically significantly vary between the different media conditions either at early or late passages. The cycle numbers were lower (indicating higher expression) for ABCG2 as compared to OCT-4.

Expression profile of pluripotency/stem cell markers. Relative levels of ABCG2 and OCT-4 mRNA in WJ-MSCs cultured in different basal media at early (P3–P5,
In immunofluorescence, Oct-4 protein expression was observed as punctate staining located in the nucleus. No differences in the subcellular localization or in the degree of expression were observed between the WJ-MSCs cultured in the different basal media either at early (Fig. 4A–4D) or late passages (Fig. 4E–4H). On the contrary, we could not detect any ABCG2 protein in the WJ-MSCs by immunostaining when cultured under the different media conditions either at early or late passages (Fig. 4N). Breast cancer cell line MCF-7 was used as a positive control (Fig. 4M). When analyzed for the expression of vimentin, an intermediate filament protein and mesenchymal marker, immunofluorescence showed that WJ-MSCs cultured in all the four different basal media strongly expressed vimentin at early (Fig. 4I–4L) as well as late passages (data not shown).
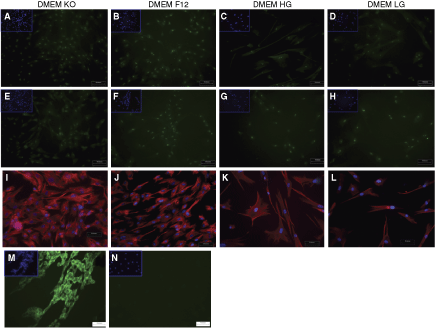
Immunofluorescence staining for pluripotency and mesenchymal markers in WJ-MSCs. Representative images of Oct-4 protein expression (shown in green) in WJ-MSC cultures grown in different media at early (P3–P5,
Karyotype analysis and transformation markers
To evaluate numerical and structural chromosomal abnormalities in WJ-MSCs after long-term expansion under different media conditions, samples were GTG-banded (Fig. 5A). None of the samples were found to be abnormal, indicating stable karyotypes through 15 passages.

(
There are reports which suggest that MSCs when cultured in vitro for a long time can undergo spontaneous transformation and immortalize at a high frequency [28 –30]. Since till 15–20 passages, our WJ-MSCs cultured in DMEM-KO and DMEM-F12 do not have many senescent cells, we decided to look for signs of transformation in our late cultures. Tumor suppressor genes are inactivated in MSC senescence and crisis bypass and DNA-repair mechanisms are instrumental for bypass of senescence and crisis. Hence, we decided to look at the mRNA levels of some tumor suppressor genes, p16, p21, and p53; DNA repair enzymes, RAD51, ERCC3, and XRCC4; and the oncogene, c-Myc by RT-PCR in our late cultures of WJ-MSCs, which were grown in DMEM-KO (Fig. 5B). No major difference was observed in the expression levels of these genes till passage 20. However, preliminary data showed a small increase in expression levels of p53, RAD51, and c-Myc by RT-PCR for one culture of WJ-MSC at passage 30 (data not shown). Since a lot of molecular alterations involving several genes and pathways lead to the transformation process, a single positive or negative control encompassing all these genes could not be included with the experiments.
Among the various basal media tested, DMEM-KO was selected for the subsequent experiments.
Clinical scale expansion of WJ-MSCs
In an attempt to propagate sufficient quantities of WJ-MSCs for clinical use, we cultured them in DMEM-KO medium in a one-cell stack (636 cm2), followed by a two- and a five-cell stack. Cell stacks are large cell culture containers used for bioproduction. When cultured in a five-cell stack, a total of ∼4.8 × 109 cells could be derived from a single cord within 5–6 passages. The expanded cells retained the immunophenotypic properties and expressed the classical MSC surface marker proteins as determined by flow cytometry though the levels of CD73, CD105, and CD166 were a little lower than the small scale cultures (Fig. 6).

Immunophenotype of clinical-scale expanded WJ-MSCs. WJ-MSCs were cultured in DMEM-KO in five-cell stack and labeled with antibodies against human antigens CD34, CD44, CD45, CD73, CD90, CD105, CD166, and HLA-DR. Open histogram indicates background signal; shaded histogram represents positive reactivity with the indicated antibody. A representative analysis of three different WJ samples is demonstrated. Abbreviations: WJ-MSC, Wharton’s jelly-derived mesenchymal stem cell; CD, cluster of differentiation.
Stem cell pluripotent array analysis and comparison between WJ and BM-MSCs
We compared the stem cell pluripotency marker profile of WJ-MSCs and BM-MSCs, both cultured in DMEM-KO, at early passages and further to evaluate the stability of the gene expression profile over the course of culture, also at late passages using a Human Stem Cell Pluripotency Low Density Array. This array includes stem cell/pluripotent markers used to characterize undifferentiated stem cells and also some early differentiation lineage markers. BM-MSC, being the most used and well-studied MSC, was used as a calibrator or control for the array data. We pooled the cDNA from four different samples of WJ and BM-MSC each and a single replica was run. In order to evaluate the transcriptional changes between the two MSCs, we focused on genes that have >2.0 fold change at either or both early and late passages as shown in Table 2. Raw data provided as supplementary data (Supplementary Table 1 and Supplementary Table 2; Supplementary materials are available online at http://www.liebertpub.com).
cDNA from four different samples (n = 4) were pooled before being used in the array.
∗Assessed by Taqman gene expression assay and not part of Human Stem Cell Pluripotency Array.
Abbreviations: BM-MSC, bone marrow-derived mesenchymal stem cell; ND, not detected (>35 cycles).
It has been previously reported in literature that MSCs isolated from WJ expand faster and to a greater extent as compared to BM-MSCs and, hence, they are considered to be a primitive stromal population [20]. Accordingly, we found an increased expression of undifferentiated human embryonic stem (hES) cell markers such as NANOG, DNMT3B, and GABRB3 and other Pluripotent/stem cell markers, like BRIX, CD9, GAL, GRB7, KIT, LIN28, NR5A2, NR6A1, and REX1 in WJ-MSCs as compared to BM-MSCs. However, prolonged in vitro culture of WJ-MSCs did result in downregulation for some of these genes such as BRIX, CD9, DNMTB3, GABRB3, GAL, KIT, and REX1. We did not detect any expression of NANOG and LIN28 at the late passages of WJ-MSCs. NR5A2 and NR6A1 showed increased expression in the late cultures of WJ-MSCs. IFITM1, IFITM2 are two stem cell markers that are strongly upregulated in BM-MSCs as compared to WJ-MSCs at the early passages. But interestingly at late passages, there is a downregulation of these two genes in the BM-MSCs, while the expression level remains almost the same in the WJ-MSCs, thus changing the shift toward a stronger expression in WJ at late passages. BM-MSCs have a higher expression of PODXL than WJ-MSCs at both early and late passages. Then, there are a few embryonic stem cell/pluripotency marker genes like OCT4 and COMMD3 for which BM-MSC and WJ-MSC share a similar expression profile while a few others like FOXD3 and SOX2 that are not detected in either of the two MSCs.
Some of the other strongly upregulated genes in WJ-MSCs were those which are involved in endoderm development like FLT1, GATA4, GATA6, ISL1, LAMA1, SOX17, and SERPINA1. Out of these, GATA4, FLT1, and SOX17 are strongly expressed in the WJ-MSCs both at early and late passages, while not detected at all in the BM-MSC cultures (Fig. 7). In case of SERPINA1, at early passages there was an increased expression in BM-MSCs as compared to WJ-MSCs, while at late passages there was a downregulation of this gene in BM-MSCs; but the level remained the same in WJ-MSCs, hence reversing the trend at the late passages. GFAP, NES, SEMA3A, which are involved in neuronal development and regulation, were also found to be upregulated in WJ-MSCs as compared to BM-MSCs at both early and late passages, though no particular trend was noted over the course of the culture.
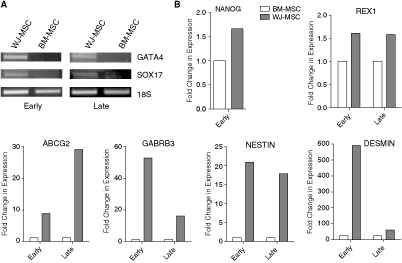
Reverse transcription polymerase chain reaction (RT-PCR) analysis of selected differentially expressed genes between WJ-MSCs and BM-MSCs at early and late passages. (
An overexpression of desmin, intermediate filament protein found in muscle cells, was found at both early and late cultures of WJ-MSCs as compared to BM-MSCs.
We found an increased expression of NOGGIN, PECAM1, SFRP2, and SYP in BM-MSCs than in WJ-MSCs both at early and late passages (Table 2).
Quantitative RT-PCR confirmed the fidelity of the array data, where select genes expressed in accordance with their differential expression pattern in the array (see Fig. 7). There was good agreement between expression by RT-PCR and the array data, though some variation in the Ct values or fold change in expression was observed as the array was based on Taqman chemistry and the run was done using ABI 7900HT instrument, while, to verify the data, quantitative RT-PCRs were done using SYBR green reagent and using an ABI 7500HT instrument.
Discussion
MSCs can differentiate to multiple lineages, induce peripheral tolerance and migrate to injured tissues, where they can inhibit the release of proinflammatory cytokines and promote the survival of damaged cells. These intriguing properties of MSCs make them strong candidates for cell-based therapy against various human diseases. MSCs can be isolated from many adult tissues but as their in vivo frequencies are intrinsically low, ex vivo expansion of MSCs is a prerequisite to obtain clinical dose for subsequent therapeutic applications. Also, we need a better understanding of the pluripotent/stem cell specific and early lineage markers that the MSCs from different tissue sources express in order to understand their self-renewal potential and differentiation propensity before introducing them to clinical applications. In this study, we compared different basal media to investigate which would promote the expansion of WJ-MSCs the best without the cells losing their plasticity or stemness. Next, comparative gene expression profiling of WJ-MSCs and BM-MSCs gave an insight into the stem cell and early lineage markers, which these two MSCs express.
Evaluation of different basal media led us to conclude that DMEM-KO and DMEM-F12 are most suitable for the expansion of WJ-MSCs as evident from population doubling time, the number of population doublings, and total cell numbers. This is in accordance with data obtained previously for characterization and upscaling of BM-MSCs from our group [25]. The WJ-MSCs failed to grow for more than 10–12 passages in DMEM-LG, had low total cell numbers and long doubling time. Hence, two conclusions can be drawn, first, perhaps low glucose concentration in medium does not support extensive proliferation of WJ-MSCs in culture, second, other supporting factors like additional amino acids, vitamins, inorganic salts, and components present in DMEM-KO and DMEM-F12 might be crucial for optimal expansion. DMEM-KO, which is optimized for growth of undifferentiated embryonic stem cells and mimics the natural environment of embryonic tissues, is a proprietary basal medium with reduced osmolality. Though the exact composition of DMEM-KO is not known, it offered the advantage of retaining differentiation potential of WJ-MSCs, especially osteogenic, till late passages over DMEM-F12. Therefore, in our study, out of the different basal media tested DMEM-KO turned out to be the best medium for the expansion as well as differentiation of WJ-MSCs. To facilitate clinical use of WJ-MSCs, we decided to expose the cells to 10% of serum and not higher concentrations. In fact, in an effort to avoid the use of FBS in our cultures, we tried supplementing DMEM-KO with 15% Knockout serum replacement (KoSR), but unfortunately KoSR did not support cell growth for WJ-MSCs (data not shown).
While the WJ-MSCs at both early and late passages expressed all the classical MSC marker proteins, the detection level for some of the markers were a little lower at late, that is, beyond 15 passages, suggesting that for clinical applications early passage cells should be preferred.
As WJ-MSCs originate from embryonic epiblast and are known to possess properties between ESCs and adult stem cells, they are thought to be more primitive than MSCs from adult tissue sources [31]. Not much is known regarding the expression of various stem cell genes in fetus versus adult-derived MSCs and their expression levels. We found the expression of Oct-4 and ABCG2 at the mRNA level and a distinct nuclear punctate staining of Oct-4 by immunofluorescence in our WJ-MSCs. Failure in finding ABCG2 staining by immunofluorescence in WJ-MSCs could possibly be explained by overexpression of miRNAs, which target ABCG2 in these cells and work in this direction is currently underway. Next, in an attempt to determine the degree of stemness exhibited by the WJ-MSCs, we used the Human Stem Cell Pluripotency array, containing markers to characterize undifferentiated stem cells and their early differentiated derivatives, that is, to screen and identify a wide range of stem cell and early lineage differentiation genes expressed by the WJ-MSCs at early and late passages and compared against early and late cultures of BM-MSCs. Based on the characterization of human ES cell lines from laboratories worldwide by the International Stem Cell Initiative (using NANOG as a reference) NANOG [32,33], POU5F [34], and TDGF [35] are associated with pluripotent state in both mES and hES cells and are regarded as archetypal pluripotent stem cell markers. NANOG, TDGF, POU5F1, GABRB3, GDF3, and DNMT3B, which are expressed in undifferentiated hES cells and show downregulation upon differentiation, are thought to constitute a core set of markers to define undifferentiated hES cells. An additional 14 genes, which are correlated to stemness (FGF4, GAL, LEFTB, IFITM1, NODAL, TERT, UTF1, FOXD3, EBAF, LIN28, GRB7, PODXL, CD9, BRIX) are also assigned to undifferentiated hES cells but could be expressed in derivatives differentiating from hES cells [36]. In our cultures of WJ-MSCs, we found a higher expression (>2-fold) of undifferentiated hES markers NANOG, DNMTB3, GABRB3, and other stemness markers like ABCG2, BRIX, CD9, GAL, GRB7, KIT, LIN28, NR5A2, NR6A1, and REX1 when compared to BM-MSCs both at early and late passages. Though within WJ samples, there was a downregulation for many of these genes upon prolonged culture. IFITM1, IFITM2, and PODXL are some stem cell markers, where WJ-MSCs had a lower expression than BM-MSCs at early passages. Though WJ-MSCs express most of the stem cell/pluripotent markers at a lower level than hESCs, the expression levels are higher than that in BM-MSCs, which would indicate a higher degree of stemness and self-renewal capacity in these cells as compared to BM-MSCs.
The Human Stem Cell Pluripotency Array (ABI) that we used included, other than the pluripotency and stem cell markers, markers associated with differentiated lineages. Another prominent difference between the WJ-MSCs and BM-MSCs was in the expression of genes associated with endoderm lineage or development. These included FLT1, GATA4, GATA6, ISL1, LAMA1, SOX17, and SERPINA1, which were strongly expressed in the WJ-MSCs. In fact, FLT1, GATA4, and SOX17 were found to be expressed only by the WJ-MSCs both at early and late passages. Again, WJ-MSCs overexpressed some transcripts associated with ectodermal lineage or neuronal development like NESTIN, GFAP, and SEMA3A suggesting that these cells might have a stronger propensity toward endodermal and neuronal differentiation as compared to BM-MSCs. In fact, lending support to our finding, in recent years there have been quite a few reports regarding the therapeutic role of WJ-MSCs in neurodegenerative disorders [37 –41].
In the present study, on culturing WJ-MSCs in DMEM-KO medium, we found short population doubling time in culture, minimal senescence by β-galactosidase staining, and no chromosomal abnormalities or expression of tumor suppressor/oncogenes, while retaining differentiation potential till late passages. It was earlier reported that MSCs enter senescence and start losing their stem cell characteristics almost undetectably from the moment in vitro cell culture begins [42]. Considering this fact, MSCs should only be used for clinical purpose at an early passage of in vitro culture, which may severely restrict the product development process. However, in the present study, WJ-MSCs exhibited both hES and hMSC markers and maintained stemness for several serial passages without exhibiting high senescence rate. This reconfirms the fact that WJ-MSCs, which are of fetal origin, have more primitive characteristics than adult MSCs and suggests that WJ-MSCs might be true stem cell populations. From a single umbilical cord, these cells could also be upscaled to a clinical dose in just 5–6 passages, though there was a small decrease in the detection level for some of the surface markers and currently, we are trying to improve upon the culture conditions for large scale expansion in our laboratory and overcome this. These properties make the Wharton’s jelly of umbilical cord a rich source of MSCs suitable for banking and universal applications, where a clinical dose of cells or repeated infusions are required without having to worry about donor site morbidity or plasticity decreasing with age. This study also provides an insight into the differential stem cell and lineage marker expression by WJ and BM derived MSCs at early and late passages. WJ-MSCs uniquely expressed some early endodermal lineage markers, and also there was upregulation of certain ectoderm/neuronal genes as compared to BM-MSCs. Thus, our observations indicate that BM and WJ-MSCs might be differently committed cells.
The WJ-MSCs could be precommitted toward endodermal and neuronal fate and further research is necessary to confirm their differentiation capacities toward these lineages. Thus, we derived gene signatures for WJ and BM-MSCs, which can be used to guide future efforts toward in vitro differentiation or cell based therapies.
Footnotes
Acknowledgments
This work was fully funded by internal funding of Stempeutics Research Pvt Ltd, Bangalore, India. We are grateful to Dr. Praveena Shenoi, Consultant Obstetrician and Gynaecologist, Manipal Hospital, for generously providing the umbilical cord samples, to Dr. Ramesh Bhonde, Stempeutics, for his expert comments on this manuscript, and Dr. Swathi Sundar Raj, Stempeutics, for her assistance in FACS data analysis and plotting of growth curves. U.N. and V.B.R. contributed equally to this work.
Author Disclosure Statement
The authors declare no competing financial interests.