
Abstract
The differentiation and/or therapeutic potential of skeletal muscle–derived stem cells for cardiac infarction have been studied extensively for use in cellular cardiomyoplasty, as injured cardiomyocytes exhibit limited regenerative capacity. We previously reported cardio-myogenic differentiation of skeletal muscle–derived CD34+/45− (Sk-34) stem cells after therapeutic transplantation. However, the clonal differentiation potential of these cells remains unknown. Here, we show that skeletal muscle–derived CD34−/45− (Sk-DN) stem cells, which are situated upstream of Sk-34 cells in the same lineage, exhibit clonal differentiation into cardiomyocytes after single cell-derived single-sphere implantation into myocardium. Sk-DN cells were enzymatically isolated from green fluorescent protein (GFP) transgenic mice and purified by flow cytometry, and were then clonally cultured in collagen-based medium with bFGF and EGF after clonal cell sorting. Single cell-derived single-sphere colonies of Sk-DN cells were directly implanted into the wild-type mouse myocardium. At 4 weeks after implantation, donor cells exhibited typical cardiomyocyte structure with the formation of gap-junctions between donor and recipient cells. Expression of specific mRNAs for cardiomyocytes, such as cardiac actin and GATA-4, Nkx2-5, Isl-1, Mef2, and Hand2, were also seen in clonal cell cultures of Sk-DN cells. Cell fusion–independent differentiation was also confirmed by bulk cell transplantation using Cre- and loxP (enhanced GFP)-mice. We conclude that Sk-DN cells can give rise to cardiac muscle cells clonally, and that skeletal muscle includes a practical cell source for cellular cardiomyoplasty.
Introduction
S
We previously demonstrated that freshly isolated murine skeletal muscle–derived CD34+/45− (Sk-34) cells are multipotent stem cells [1,2], and that they can give rise to cardiomyocytes having desmosomes and intercalated discs associated with gap-junctions after transplantation into the myocardial infarction (MI) zone, significantly contributing to the functional recovery of the left ventricle (LV) [3]. Differentiation into cardiomyocytes was also confirmed by FISH analysis and in vitro coculture with embryonic cardiomyocytes [3]. However, clonal differentiation potential has yet to be defined in Sk-34 cells. Confirmation of this potential is vitally important for stem cell and developmental biology, and would allow the establishment of a practical cell source of future cardiomyoplasty.
We also identified a further cell population in the CD34−/CD45− (Sk-DN) fraction as putative immature stem cells that typically form sphere-like colonies having high colony-forming activity (over 40%) in a collagen-based semisolid cell culture system with bFGF and EGF [4]. Sk-DN cells also exhibited myogenic-endothelial differentiation capacity during culture [4] and high therapeutic potential for muscle-nerve-blood vessel units [5], as similarly reported for Sk-34 cells [2]. In addition, the characteristic single cell-derived sphere-colony formation in Sk-DN cells is suitable for clonal differentiation analysis [6]. We then found that Sk-DN cells are highly immature stem cells situated upstream of Sk-34 cells in the same lineage, and that they are potentially capable of self-renewal [7].
Based on the relationship between Sk-34 and Sk-DN cells in the same lineage and the basic clonal sphere-colony forming potential of Sk-DN cells, we performed clonal (single cell-derived single-sphere) transplantation of Sk-DN cells into heart muscle. In vivo differentiation was confirmed by immunohistochemical detection of gap-junctions between implanted donor cells and recipient cardiac muscle cells associated with cardiac troponin expression. Single-cell and clonal 2 to 40 cell, single-sphere, and bulk cell RT-PCR were then performed in order to analyze the expression of cardiac, skeletal, and smooth muscle-specific mRNAs.
Materials and Methods
Animals
Green fluorescent protein transgenic mice (GFP-Tg mice; C57BL/6 TgN[act EGFP]Osb Y01, provided by Dr. M. Okabe, Osaka University, Osaka, Japan) [8] were used as donor mice in cell transplantation and culture studies. C57BL/6N wild-type mice were used as recipients in clonal implantation experiments. Cre- and loxP-mice [B6. Cg-Tg (CAG-cre, CZ-M020sc; BRC 01828) and (CAG-floxed Neo-EGFP, REP080sb; BRC 02096)] provided by RIKEN BRC, which are participating in the National Bio-Resource Project of MEXT, Japan, were used for confirmation studies of cell fusion after transplantation. All experimental procedures were conducted in accordance with the Japanese Physiological Society Guidelines for the Care and Use of Laboratory Animals, as approved by the Tokai University School of Medicine Committee on Animal Care and Use.
Cell purification
Whole muscles from the thigh and lower leg (tibialis anterior, extensor digitorum longus, soleus, plantaris, gastrocnemius, and quadriceps femoris) of 3- to 8-week-old GFP-Tg mice were treated with 0.1% collagenase type IA (Sigma-Aldrich, St. Louis, MO) in Dulbecco’s modified Eagle’s medium (DMEM) containing 5%–10% fetal calf serum (FCS) with gentle agitation for 2 h at 37°C, and the interstitial cells were extracted. Extracted cells were filtered through 70-µm, 40-µm, and 20-µm nylon strainers in order to remove muscle fibers and other debris, and were then washed and resuspended in Iscove’s modified Dulbecco’s medium (IMDM) containing 10% FCS, yielding enzymatically extracted cells. These cells were stained with allophycocyanin (APC)-conjugated antimouse CD34 (RAM34, 17-0341; eBioscience, San Diego, CA) and pacific blue-conjugated antimouse CD45 (30-F11, 103126; BioLegend, San Diego, CA). CD34−/CD45− (Sk-DN) cells were then collected. Live cells were counted after cells positive for propidium iodide (PI) were excluded as dead cells. Cell analysis and sorting were carried out on a FACSAria (Becton Dickinson Japan, Tokyo).
Clonal cell implantation and bulk cell transplantation
Clonal analysis for Sk-DN cells were performed based on the typical characteristics of sphere-like colony formation from single cells. One hundred freshly isolated Sk-DN cells were clone-sorted into two 96-well plates (50 cells each) by FACS and individually cultured in collagen-based semisolid medium (CollagenCult H4742; StemCell Tech, Vancouver, Canada) with 10 ng/mL bFGF and 20 ng/mL EGF. Two weeks after culture, individual colony-forming units (CFUs) were suspended in 0.5% collagenase/phosphate buffered saline (PBS) for 5 min at 37°C, were washed with serum-free medium, and were prepared for clonal cell implantation.
For clonal implantation, typical single spheres (see Fig. 1A) from GFP-Tg mice were randomly selected and directly implanted into the left ventricular (LV) wall of C57BL/6 wild-type mice (n equals; 6), using a fine grass micropipette without infarction, in order to fix the injected cells in a limited area. This method enabled relatively easy detection of engrafted cells at 4 weeks after transplantation.
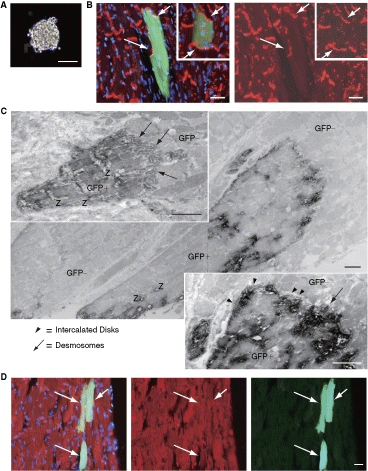
Histological characteristics at 4 weeks after clonal single sphere implantation of skeletal muscle–derived CD34−/45− cells. (
In order to confirm whether spontaneous cell fusion occurs, we performed bulk cell transplantation with three different donor/recipient combinations: (1) Cre/loxp mice (n = 5); (2) loxP/Cre mice (n = 5); and (3) GFP-Tg/wild-type mice (positive control of Cre-loxP system cell transplantation, n = 5). In this bulk cell transplantation, an MI model was produced in recipient mouse LV under halothane anesthesia (Fluothane, Takeda Chemical, Osaka, Japan). We have previously observed vigorous engraftment of transplanted Sk-34 cells (situated hierarchically downstream of the present Sk-DN cells in the same lineage) in the MI region, even in heterografts (GFP-mouse to nude rat) [3]. After tracheal insertion and initiation of ventilation (room air, rate 60 cycles/min, tidal volume 1 mL/100 g body weight, Harvard Apparatus Rodent Ventricular, model 683), the heart was exposed by left thoracotomy, and the proximal left coronary artery was ligated. Sk-DN cells were obtained from donor mouse skeletal muscles, were cultured in same collagen-based medium for 6 days, and were then suspended with collagenase. After washing with nonserum DMEM, cells were transplanted directly into the infarcted area of the LV wall in recipient mice (5 × 105 cells/animal).
RT-PCR
Single-cell RT-PCR was performed based on a highly optimized global RT-PCR procedure [9,10]. Freshly isolated Sk-DN cells were clonally cultured in collagen-based semisolid medium (StemCell Tech, see above). Single cells and/or clones composed of 2–8 cells were obtained after 3 days and clonal >40 cells were obtained after 5 days of clonal cell culture. Cells were then manually removed with a fine-tip micropipette, and suspended/washed in cold RNase-free 0.01 M PBS. Samples were lyzed with 9 µL of cold lysis–first-strand synthesis solution containing first-strand buffer (Invitrogen, Carlsbad, CA), 1% NP-40, 1 mM dithiothreitol, 0.01 mM dNTPs, 3.4 nM dT30-containing primer (AAGCAGTGGTATCAACGCAGAGTGGCCATTACGGCCG TACTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT) and RNase inhibitors (Ambion, Austin, TX; Eppendorf, Hamburg, Germany). Samples were quickly frozen with liquid nitrogen and were stored at −80°C until use. For analysis, samples were heated at 65°C for 5 min and placed on ice. Lysates were equally divided into 2 PCR tubes, to which 100 U of SuperScript III reverse transcriptase (Invitrogen) or 0.5µL of nuclease-free water (negative control) was added.
The first cDNA strand was synthesized by incubation for 60 min at 45°C. The reaction was stopped by heating at 65°C for 10 min. After cooling on ice, 1.5 µL of 1 U RNase H solution (Invitrogen), 0.5 µL of 75 mM MgCl2 and 0.5 µL of nuclease-free water were added to the test sample. RNA was degraded by incubation for 15 min at 37°C and RNase H was inactivated for 10 min at 65°C. Samples were immediately cooled on ice and 6.5 µL of 2X poly-dA tailing solution containing 2X terminal deoxynucleotidyl transferase buffer, 3 mM CoCl2, 1.5 mM dATP, and 15 U of terminal deoxynucleotidyl transferase (Promega, Madison, WI) were added to 6.5 µL of first-strand cDNA solution. Poly-dA tailing was performed for 15 min at 37°C and was stopped by heating at 65°C for 10 min. Poly-dA tailed cDNA was preamplified using a sequence nonspecific 2-step PCR protocol with dT30-containing primer. Preamplification was performed in a 20-µL volume containing ExTaq buffer, 1 U of ExTaq-polymerase (Takara Bio, Shiga, Japan), 8.3 µM dT30-containing primer, 0.65 mM dNTPs and 4 µL of poly dA tailed cDNA.
The second strand was synthesized by incubating for 1 min at 94°C, 2 min at 50°C, and 2 min at 72°C, followed by 35 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 2 min. Duplicate reaction tubes were employed in order to compensate for experimental errors. The reaction products from the 2 tubes were mixed and this mixture was used as a template for the second preamplification. The second PCR was performed in a 20-µL volume containing ExTaq buffer, 1 U of ExTaq-polymerase, 2 µM dT30-containing primer, 0.2 mM dNTPs, and 2 µL of the first PCR product. The reaction comprised 35 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 2 min.
The second PCR product was subjected to 2% agarose gel electrophoresis and was confirmed as a smear at 500–700 bp. Specific PCR (30 cycles of 30 s at 94°C, 30 s at 60°C, and 2 min at 72°C) was performed in a 15-µL volume containing Ex-Taq buffer, 0.8 U of ExTaq-polymerase, 0.7 µM specific sense and antisense primers, 0.2 mM dNTPs, and 0.2 µL of the second PCR product. Preparations without reverse transcriptase served as negative controls in cDNA synthesis, preamplification and specific PCR. Amplification of genomic DNA was not detected in specific PCR preparations lacking reverse transcriptase. Specific primers are summarized in Table 1.
S
In addition, to test the expression of specific markers in cultured total Sk-DN cells, bulk cell RT-PCR was performed. Sk-DN cells were cultured in the same collagen-based medium with bFGF and EGF for 4, 6, and 15 days, and were then lyzed, after which total RNA was purified using a QIAGEN RNeasy micro kit. Analysis was performed using the same protocol described above.
Immunostaining and immunoelectron microscopy
Transplanted hearts were perfused with 0.01 M PBS and were fixed overnight in 4% paraformaldehyde/0.1 M phosphate buffer (4% PFA/PB). After washing with a graded sucrose (0%–25%)/0.01 M PBS series, hearts were quick-frozen in isopentane, and several 7-µm cross-sections were obtained. Localization of gap-junctions was detected using rabbit anti-connexin43 polyclonal antibody (c 6219; dilution: 1:1,000; incubation: 4°C overnight; Sigma, St. Louis, MO). Differentiation into cardiac muscle cells was also confirmed by immunostaining of cardiac troponin T (goat anticardiac Troponin T polyclonal antibody, sc-8121; dilution: 1:100; incubation: 4°C overnight; Santa Cruz Biotechnology, Santa Cruz, CA). Reactions were visualized using Alexa Fluor-594-conjugated goat antirabbit (A11037) and donkey antigoat antibodies (A11058; 1:500, room temp., 2 h; Molecular Probes, OR). Nuclei were counter-stained with DAPI (4,6-diamino-2-phenylindole).
For immunoelectron microscopy, sections were stained using rabbit anti-GFP antibody (A11122; 1:300, 4°C overnight; Molecular Probes) and HRP-conjugated antirabbit antibody (1:200, 4°C overnight; Dako, Carpinteria, CA). Reactions were visualized with DAB after fixation in 1% glutaraldehyde/0.1 M PB. Sections were then fixed in 1% osmium tetroxide/0.05 M PB, and were prepared for electron microscopic analysis.
Results
Clonal cell transplantation
In order to confirm clonal differentiation potential in vivo, single spheres (Fig. 1A) derived from single Sk-DN cells of GFP-Tg mice were directly implanted into the LV wall of C57BL/6 wild-type mice without infarction. This type of sphere colony was typically composed of around 100 cells. At 4 weeks after implantation, GFP+ cells having gap-junctions (positive for gap-junction alpha-1 protein; connexin43, red reactions) among GFP− (host) cardiac muscle cells were evident in the host LV wall (Fig. 1B, arrows). Implanted GFP+ donor cells also exhibited the formation of intercalated disks (arrowheads in Fig. 1C) including desmosomes (arrows in Fig. 1C) and expression of cardiac troponin C (Fig. 1D, arrows), indicating complete differentiation into cardiomyocytes. The approximate number of engrafted donor-derived cardiomyocytes and/or cardiomyocyte-like cells was 10–30 cells. Other cell types, such as vascular cells and multinucleated skeletal muscle cells, were not detected after clonal implantation. The same trend was obtained in 3 of 6 mice (50%). Thus, it is possible that skeletal muscle–derived Sk-DN cells clonally give rise to cardiomyocytes at a rate of 50% after in vivo transplantation.
Interestingly, GFP+ primitive and relatively mature cardiomyocytes were also evident after clonal transplantation under immunoelectron microscopy (Fig. 2A). Primitive cardiomyocytes had slight and immature contractile filaments in the periphery (arrows in Fig. 2B, 2C), in contrast to the regular appearance of Z-disks in the relatively mature cardiomyocyte (situated left-side in Fig. 2A). The typical structure of intercalated disks, desmosomes, and gap-junctions shown in Figure 1 was not observed in these cells. Higher magnification images of Figure 1C (immunoelectron micrographs) is available in the online Supplementary Figure 1. (Supplementary materials are available online at http://www.liebertpub.com/)
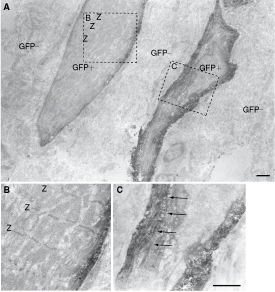
Immunoelectron micrograph of donor cell-derived primitive cardiomyocytes after clonal transplantation. Green fluorescent protein (having black dots) donor-derived cell shows similar shape as cardiomyocytes having immature contractile filaments in the periphery (arrows in
RT-PCR for single, clonal, and bulk cells after culture
The cardiomyogenic commitment of Sk-DN cells was further analyzed by performing RT-PCR analysis for single cells, and clonally divided 2 to 8 and >40 cells (spheres) after 3 and 6 days of culture (Fig. 3A). In this analysis, “single cell” refers to the stage before cell division, “2 cells” refers to the stage after the first cell division, “3–4 cells” refers to the second cell division, “5–8 cells” refers to the third division, and “>40 cells” and/or “sphere” refers to more than 6 divisions. The expression of 3 types of muscle-related markers for skeletal (red), smooth (purple), and cardiac (black) was observed at the single-cell stage, but the appearance ratio was low and the cardiac lineage marker expression was clearly lower than that for skeletal and smooth muscle lineage (Fig. 3B). This trend was basically maintained with further cellular divisions, and the highest ratio was observed at the >40 cell stage (small sphere). This indicates that specific marker expression was accelerated following the increase in cell number in each colony, and this trend was more apparent in cardiac and vascular cell lineage markers. Thus, it was clear that expression of specific marker mRNAs was relatively weak in the vascular and cardiac cell lineage when compared with the skeletal and smooth muscle lineage. Supporting the results for single and clonal cell RT-PCR, expression of specific mRNAs was stronger in cultured bulk Sk-DN cells basically composed of numerous sphere colonies, and all markers were detected from 4 to 15 days of culture (Fig. 4).
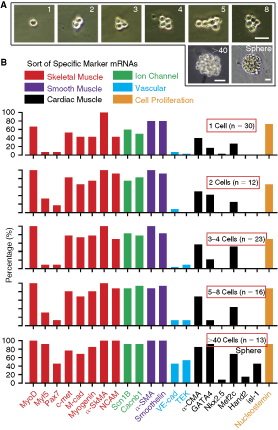
Characteristics of single and clonal 2, 3–4, 5–8, and >40 cells. (
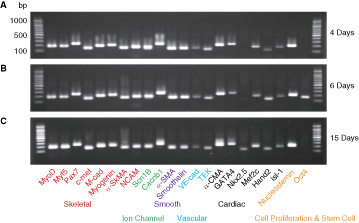
RT-PCR for cultured bulk skeletal muscle–derived CD34−/45− cells. Sk-DN cells were cultured for 4 days (
Bulk cell transplantation
In order to clarify the spontaneous cell fusion mechanism, we also performed 3 sets of bulk Sk-DN cell transplantations (allograft) using different combinations of donors and recipients (Cre- and loxP-EGFP mice), in addition to GFP-Tg/wild-type mice as a positive control (see Materials and Methods). At 4 weeks after transplantation, there were no EGFP-positive cells found in either recipient Cre- or loxP-mouse LV wall, whereas large numbers of GFP+ cardiac-like cells positive and/or weekly positive for cardiac troponin-T were seen in wild-type recipient mouse LV wall (Fig. 5A and B). Large numbers of engrafted GFP+ donor cells were evident on longitudinal (A) and cross-sectional (B) dimensions. These cells were detected at ∼150–300 cells/section (7 µm), and serially appeared at around 2 mm in depth. Interestingly, engrafted Sk-DN cells filled in the MI zone, but reactions for anticardiac troponin-T were stronger in the border zone (arrows in A and B) than in the central zone. This phenomenon suggests that differentiation of engrafted Sk-DN cells into cardiomyocytes began at the border zones and spread to the central MI zone. These results indicate that cultured and expanded bulk Sk-DN cells are actively engrafted into recipient cardiac tissues, but that no spontaneous cell fusions occur.
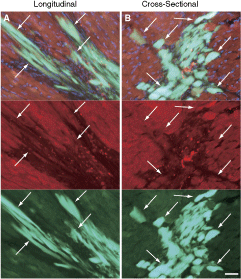
Histological observation at 4 weeks after bulk skeletal muscle–derived CD34−/45− cell transplantation. There were no green fluorescent protein (GFP) cells found in Cre-loxP system transplantation, thus suggesting that no cell fusions occurred. However, vigorous cellular engraftment was seen in GFP-Tg-wild-type mouse transplantation, both in longitudinal (
Discussion
The clonal differentiation potential of skeletal muscle–derived clonal Sk-DN cells was examined by immunohistochemistry and immunoelectron microscopy after clonal single sphere transplantation into a cardiac muscle environment without cardiac infarction. Based on the results shown in Figures 1 and 2, transplanted clonal Sk-DN cell showed 3 different cellular stages of cardiac differentiation; immature, relatively mature, and completely mature. This suggests that transplanted Sk-DN cells undergo the cardiac differentiation process. Transplanted cells in clonal spheres were susceptible to the effects of the recipient cardiac muscle microenvironment via paracrine cytokines and the physical dynamic motion of the LV wall, as transplanted cells were in close proximity (see Fig. 1). Factors from microinjection (glass pipette) inducing micro tissue damage and calcium signals [12] may have also been a strong inducer. As a result, endogenous cardiac lineage potential (eg, expression of cardiac lineage marker mRNAs) in transplanted cells reacts to inductive signals provided by the new microenvironment, and the transplanted cells gradually differentiate into complete cardiomyocytes via the acquisition of cell phenotypes endogenous to the new microenvironment [13,14]. However, the elimination of possible spontaneous cell fusion mechanisms is necessary for this type of cell transplantation study. For this purpose, cultured bulk Sk-DN cell transplantation using Cre- and loxP- (EGFP) mice demonstrated that there were no cell fusions, whereas large numbers of GFP+ cells were seen after the same bulk-cell transplantation from GFP-Tg mice to wild-type mice (Fig. 5). In bulk-cell transplantation, it is likely that topological effects of the microenvironment are stronger in the border zone of the MI than that in the central zone, as shown by the reaction of anticardiac troponin-T in Figure 5. Thus, we conclude that the present differentiation of Sk-DN cells into cardiomyocytes was not dependent on spontaneous cell fusion, but was dependent on the cellular differentiation mechanisms of stem cells.
We also reported that freshly isolated Sk-34 cells can give rise to cardiomyocytes having desmosomes and intercalated discs associated with gap-junctions after transplantation into the MI zone, significantly contributing to functional recovery of the LV [3]. In addition, we demonstrated that Sk-DN cells are enriched multipotent stem cells situated upstream of Sk-34 cells in the same lineage, and are potentially capable of self-renewal [7]. Thus, it is possible that the previous cardiac differentiation of Sk-34 cells after therapeutic bulk-cells transplantation and its significant contribution to functional recovery after MI depended on the clonal differentiation potential of this stem cell lineage. However, it is likely that the present Sk-DN and previously reported Sk-34 cells may default to differentiation into skeletal muscle, vascular, and peripheral nerve-related cells. Because when these cells were transplanted into relatively neutral conditions in vivo, such as beneath the kidney capsule, they differentiated into skeletal muscle fibers with capillary and Schwann cells [2,5].
For the expression of specific mRNAs during cell culture, cell number-dependent increases in expression ratio were observed, particularly in the vascular and cardiac cell lineages (Fig. 3), and almost all expressions were observed in the bulk-cell culture (Fig. 4). These results suggest the importance of cellular interactions. In other words, specific marker expression was induced and/or accelerated by cell-to-cell interactions in each sphere colony, and sphere formation is beneficial for maintaining cellular interactions and differentiation potential, as Sk-DN cells formed sphere colonies and increased their cell numbers under collagen-based cell culture conditions, even in the case of bulk cell culture [5,7].
In a previous study, Sk-34 cells also formed spheres associated with gap-junctions in the coculture of embryonic cardiomyocytes and showed spontaneous and synchronous contractions [3]. Therefore, in order to further confirm cell–cell interactions in the cultured Sk-DN cells on the basis of forming gap-junctions, we cultured Sk-DN cells with the addition of gap-junction inhibitor (18-beta-Glycyrrhetinic acid; final concentration, 75 µM) [11] for 7 days in the same manner as in the bulk cell RT-PCR experiment. Expression patterns of mRNA markers corresponded to the patterns seen in 6-day standard culture (Fig. 4); thus, the expression of cardiac-specific mRNAs was not affected by the addition of gap-junction inhibitor in the solo culture of Sk-DN cells (online Supplementary Fig. 2). These data support the notion that our reported Sk-34 cells and the present Sk-DN cells have multi-myogenic potential that can give rise to skeletal, smooth [2,5], and cardiac muscle cells [3].
Clonal differentiation into cardiomyocytes has been reported in cardiac muscle–derived stem cells [15,16]. However, to our knowledge, this is the first report to confirm the clonal differentiation potential of adult tissue-specific stem cells into cardiomyocytes. These Sk-34 and Sk-DN cells also showed differentiation into mesodermal (skeletal muscle cells, pericytes, vascular smooth muscle cells, endothelial cells, and adipocytes) and ectodermal (Schwann cells and perineurium) cell lineages after in vivo transplantation into damaged skeletal muscle [2,5]. This mesodermal–ectodermal lineage differentiation was also clonally confirmed using Sk-DN spheres [6], similar to the present study. This type of multipotency in Sk-DN (and/or previous Sk-34) cells is of interest in the fields of cell and developmental biology. The origins of locally preserved adult stem cells have been extensively reviewed by Young and Black [17] and Young et al. [18]. Based on their classification, cells that are able to differentiate into ectodermal and mesodermal lineages, including vascular-related cells, are epiblastic-like stem cells situated just under the inner-cell mass during mammalian embryonic development. Cardiac-related cells can be derived from the lateral plate mesoderm and/or neural crest. Thus, we speculate that the present Sk-DN cells can be categorized as epiblastic-like stem cells. These highly immature cells are probably residual cells from embryonic developmental stages and are preserved in the interstitial spaces of skeletal muscle, even after birth, as a cellular reserve [17,18]. The fact that Sk-34 and Sk-DN cells are not derived from bone marrow [2] supports this notion. The notion that residual cells from embryonic developmental stages are preserved in the interstitium can also be applied to other tissue-specific stem cells residing in the adipose tissue [19] and dermis [20,21].
It has been reported that adult murine skeletal muscle contains cells that can differentiate into cardiomyocytes in vitro, and these are referred to as Spoc cells (skeletal-based precursors of cardiomyocytes) [22]. Spoc cells exhibit CD34−/CD45−/C-kit−/Sca-1− at initial isolation, and show round shape, floating and/or weakly attached behavior, sphere-colony formation and spontaneous beating (contracting) in culture, and may be different from satellite cells. These in vitro characteristics correspond to the reported characteristics of both Sk-34 [1,3,7] and Sk-DN cells [4,7], although Sk-34 cells are CD34+ and mostly Sca-1+, while Sk-DN cells are CD34−/CD45−/C-kit− and include both Sca-1− (75%) and Sca-1+ (25%) fractions [4,7]. Thus, it is possible that Spoc cells represent a similar cell fraction to Sk-DN/Sca-1− cells. However, Sk-34 and Sk-DN cells are of the same lineage and the number of Sk-DN cells is small when compared to Sk-34 cells (1:16∼20) at initial isolation [1,4,7]; thus, our method showed >20-fold higher efficiency for isolation of potential cardiomyocyte precursors from skeletal muscle over Spoc cells. Furthermore, in vitro differentiation into cardiac, skeletal muscle, and neural lineages has also been reported for skeletal muscle–derived nonadherent (floating) stem cells [23]. Non- and/or less-adherent floating characteristics are also typical of Sk-DN and Sk-34 cells [1,4]. Thus, it is likely that skeletal muscle includes putative cardiac stem and/or progenitor cells.
Concluding Remarks
In conclusion, the present results clearly indicate that skeletal muscle–derived Sk-DN cells can give rise to cardiomyocytes after clonal transplantation in vivo. This is also supported by the previously reported therapeutic potential of Sk-34 cells that can give rise to cardiac muscle cells with significant functional recovery of LV function. Therefore, skeletal muscle–derived Sk-DN and Sk-34 cells represent a potential cardiomyogenic stem cell source for autologous transplantation cellular cardiomyoplasty.
Footnotes
Acknowledgments
This work was supported by a grant from the New Energy and Industrial Technology Development Organization of Japan (T.T.), and Tokai University Research aid (T.T.).
Author Disclosure Statement
The authors declare that they have no competing financial interests.