
Abstract
Human embryonic stem cell-derived neural stem cells (hESC-NSCs) are an attractive cell type for studying aspects of brain development and pathology. To develop the full potential of this model system, it is important to establish a reliable methodology for the manipulation of gene expression in hNSCs. To address this issue, we used an adenoviral vector with a CMV promoter-driven green fluorescent protein (GFP) reporter gene (Ad5-GFP). We optimized conditions for Ad5-GFP infection and assessed the efficiency of infection of whole and dissociated embryonic stem cell (ESC)-derived neurospheres as well as the effect of adenoviral vectors on cell surface marker expression, proliferation, and differentiation potential. Our results demonstrate that most neurosphere cells (∼70%) express the coxsackie and adenovirus receptor and can be infected with Ad5. More specifically, the CD133+ hESC-NSC population could be infected more efficiently than the CD133 population and both populations expressed GFP at high levels. At low multiplicity of infection (MOI < 25), the virus had no significant effect on stem cell marker expression (CD133 and Nestin), cell survival, cell proliferation rate, or differentiation potential. This model system provides a practical new approach to study human NSC function in the context of neurodegenerative and neoplastic disorders.
Introduction
T
NSCs have been isolated from neural tissue at different stages of development [2 –5], and have been generated from human mesenchymal stem cells [6] and embryonic stem cells [7 –10]. Human embryonic stem cell-derived neural stem cells (hESC-NSCs), in particular, are an attractive source of NSCs of consistent quality and quantity. Additionally, the transcriptional and functional properties of hESC-NSCs suggest that they are well suited for modeling early neural development and pathology in vitro [11,12].
Gene expression in NSCs has been manipulated using a variety of techniques such as liposomes, biolistic transfection, electroporation, and viral-mediated transfers (reviewed in Jandial, 2008 [13]). Modified versions of the human adenovirus serotype 5 (Ad5) have been used extensively for mammalian gene transfer experiments. Ad5 vectors can carry large transgenes typically driven by Rous sarcoma virus or cytomegalovirus promoters and lack open-reading frames E1/E3 necessary for viral replication in human cells. Recombinant adenoviruses of this type are well tolerated by most cells and are carried episomally within the nucleus, thus avoiding problems associated with viral integration into the host genome [14]. Adenoviral infection of rodent fetal or embryonic stem cell-derived NSCs has been demonstrated by several groups [15 –20]. However, the data are conflicting in regard to adenovirus infection of murine adult NSCs, with one report describing good infection rates [17], while a separate study using very similar methodology was unable to demonstrate significant virus uptake even at high multiplicities of infection (MOI) [21]. Although adenovirus-mediated transgene expression in human NSCs has been demonstrated in several studies [22 –24], the methodology varies greatly and the effects of adenovirus infection on adult and fetal human NSCs have not been comprehensively investigated. In order to use hESC-NSCs as a model system for genetic studies, it is important to establish a reliable infection protocol that generates reproducible data. In the present study, we optimized infection of hESC-hNSCs using an Ad5 carrying a CMV promoter-driven GFP reporter gene (AdV-GFP). We have shown that hESC-NSCs infect efficiently and we have established assays for investigating cell surface and apoptosis markers, and proliferation and differentiation after infection. Using our optimized protocols, we were able to achieve high infection rates within a viral concentration range where side effects on NSC properties were negligible.
Materials and Methods
Cell culture and neurosphere maintenance
Human neurospheres were derived from human embryonic stem cell lines hES2, hES3 (ES Cell International, Singapore, http://www.escellinternational.com) and MEL-1 (Australian Stem Cell Centre, Melbourne, Australia, http://www.stemcellcentre.edu.au) using the protocol described previously [7,8]. In brief, hESCs were co-cultured for 14 days on a layer of mitomycin-C-treated mouse embryo fibroblasts in the presence of the bone morphogenetic protein antagonist, Noggin (500 ng/mL; R&D Systems—Bioscientific, Gymes, Australia). Cell clumps were mechanically excised from Noggin-treated colonies and transferred to 96-well low adherence plates (Corning, Melbourne, Australia) where they were maintained as neurospheres. Subsequently, neurospheres were short-term cultured up to 12 weeks in neural basal medium (Gibco; Mt Waverly, Australia) supplemented with N2 (Gibco), B27 (Gibco), penicillin/streptomycin (Gibco),
Adenovirus and infection protocol
Replication-deficient adenovirus serotype 5 (Ad5) constructs (kindly provided by Dr. Bruno Meloni and Dr. Sherif Boulos, Australian Neuromuscular Research Institute, Perth, Australia) expressing enhanced green fluorescent protein (GFP) driven by a CMV promoter were used for all experiments [25]. The diameter of the neurospheres was measured and neurospheres were lysed and counted using the NucleoCounter® (Chemometec, Allerod, Denmark). A standard curve for sphere volume and cell number was generated for infecting whole neurospheres. The rest of the neurospheres were dissociated using the papain-neural tissue dissociation kit (Miltenyi Biotec, North Ryde, Australia), following the manufacturer’s protocol. In brief, cells were washed in 1× PBS (pH 7.2) and incubated for 20 min at 37°C in enzyme mix 1, and for 10 min after adding enzyme mix 2. The latter step was repeated with a further 5-min incubation and cells were triturated until no more clumps were visible. The suspension was filtered (30 µm; BD Falcon, North Ryde, Australia) and cells were washed twice with PBS. The 105 cells/well were seeded in 96-well plates (Corning, Melbourne, Australia) and incubated overnight for recovery. Whole spheres and single cells were infected using 0.1–100 virus particles per cell. GFP expression was examined 3–4 days after infection with a Zeiss Axiovert 125 inverted microscope.
Antibodies
The primary antibodies used in this study included anti-βIII-tubulin (Tuj-1, mIgG2a; R&D Systems—Bioscientific, Gymes, Australia), anti-CNPase (mIgG1; Chemicon, Kilsyth, Australia), anti-MAP2ab (mIgG1; NeoMarkers, Melbourne, Australia), anti-S100 (mIgG2a; Chemicon, Kilsyth, Australia), rabbit-anti-GFAP (rabbit IgG; DakoCytomation, Kingsgrove, Australia), anti-GFAP (mIgG1; R&D Systems—Bioscientific, Gymes, Australia), O4 (IgM; Chemicon, Boronia, Australia), CD133/2-PE (Miltenyi Biotec, North Ryde, Australia), anti-CAR (mIgG1; Abcam, Sapphire Bioscience, Redfern, Australia), anti-Nestin (Abcam—Sapphire Bioscience, Redfern, Australia). All secondary antibodies were sourced from Molecular Probes (Invitrogen, Mt. Waverly, Australia) including goat anti-mouse IgG1—AlexaFluor647, goat anti-mouse IgG2a—PE, goat anti-mouse IgG1—PE, goat anti-mouse IgM—AlexaFluor647, goat anti-mouse IgG2a—AlexaFluor555, goat anti-mouse IgG1—AlexaFluor647, goat anti-rabbit IgG—AlexaFluor546.
Flow cytometry
For flow cytometric analysis, whole neurospheres were dissociated using the trypsin-neural tissue dissociation kit (Miltenyi Biotec, North Ryde, Australia) following the protocol described previously. Reformed cell aggregates harvested after infection of already dissociated spheres were washed with PBS and incubated with TrypLE Express (1:2; Gibco, Mt. Waverly, Australia) for 5–10 min at 37°C. Cells were washed twice with cold PBS and resuspended in FACS buffer (1× PBS supplemented with 0.5% BSA (Sigma, Castle Hill, Australia) and 2 mM EDTA)
For surface antigen analysis, cells were blocked in Fc block (Miltenyi Biotec, North Ryde, Australia) for 3 min at room temperature. Primary antibodies O4, CD133/2-PE, and CAR were added and cells were incubated on ice for 20 min. Negative control cells were exposed to IgG2b-PE, IgG1-FITC (both from Santa Cruz, Biolab, Clayton, Australia), or primary antibody was omitted. Excess antibody was washed off and antibodies were conjugated with secondary antibodies. For intracellular staining, cells were first fixed in 4% paraformaldehyde (Merck, Kilsyth, Australia) for 30 min at RT and then permeabilized and blocked in 1× PBS supplemented with 10% goat serum (Zymed—Invitrogen, Mt Waverly, Australia) 0.5% BSA, 2 mM EDTA, and 0.05% Triton X-100 (Sigma, Castle Hill, Australia) for 10 min. Cells were incubated with anti-Nestin, anti-βIII-tubulin, anti-CNPase, anti-MAP2ab, anti-S100, and anti-GFAP (mIgG1) for 30 min at 4°C, washed, and primary antibody was conjugated using goat anti-mouse secondary antibodies. Excess antibody was removed and samples were analyzed with the LSR2 flow cytometer (BD, North Ryde, Australia).
Immunocytochemistry
Populated coverslips were harvested and wells were washed twice with 1× PBS and fixed for 1 h with 4% Paraformaldehyde (Merck, Kilsyth, Australia). Cells were washed three times with 1× PBS containing 1% BSA and 2 mM EDTA. For O4 staining, cells were blocked with 10% goat serum (Zymed—Invitrogen, Mt. Waverly, Australia) for 45 min and incubated with O4 overnight. Primary antibody was conjugated with secondary antibody for 60 min and cell nuclei were counterstained with Hoechst 33342 (10 µg/mL, Molecular Probes—Invitrogen, Mt. Waverly, Australia) for 10 s. For intracellular stains, cells were permeabilized and blocked using 1× PBS, 0.3% Triton X-100 (Sigma, Castle Hill, Australia), 1% BSA (Sigma, Castle Hill, Australia), and 10% normal goat serum (Zymed—Invitrogen, Mt. Waverly, Australia) for 45 min. Cells were incubated with anti-βIII-tubulin, anti-CNPase, anti-MAP2ab, anti-S100, and rabbit anti-GFAP overnight at 4°C. Excess antibody was washed off and antibodies were conjugated using appropriate secondary antibody. Cells were washed twice with PBS and once with distilled water before counterstaining with Hoechst 33342 and viewing with an inverted Zeiss Axiovert125. Images were captured with an FII camera (Olympus, Mt. Waverly, Australia) and LifeVision software (Olympus, Mt. Waverly, Australia).
Thymidine-incorporation assay
Neurospheres were dissociated using the neural tissue dissociation kit (papain, Miltenyi Biotec, North Ryde, Australia) as described earlier and cells were infected with AdV-GFP 24 h after dissociation. Tritiated thymidine (Amersham, Sydney, Australia) was added to each well (days 0, 1, 2, 3, 5, 7, and 14). After 6 h of incubation (37°C, 3% CO2), plates were frozen at −20°C. Cells were thawed and harvested onto filter mats, and radiation was measured using the MicroBeta (PerkinElmer, Melbourne, Australia). Each assay was set up in quadruplicate and an average background reading based on the media control wells was subtracted from the readings for the test samples at each time point. Proliferation was measured as average fold increase in thymidine incorporation relative to day 0.
AnnexinV-apoptosis assay
Neurospheres were dissociated as described earlier and seeded into 96-well plates (105 per well). Cells were infected with AdV-GFP and harvested 3 days post-infection. Cells were washed and stained for CD133 as previously described and AnnexinV was added following the instructions of the Vybrant Apoptosis Assay kit #6 (Invitrogen, Mt. Waverly, Australia). For dead cell discrimination SytoxRed (Invitrogen, Mt. Waverly, Australia) was added 15 min prior to analysis with the FACS Aria I (BD, North Ryde, Australia).
In vitro differentiation
For microscopy, coverslips (13 mm; ProSciTech, Thuringowa, Australia) were coated with poly-
Results
hESC-NSC populations express the coxsackie and adenovirus receptor (CAR), CD133, and Nestin
Using flow cytometry, we analyzed hESC-derived neurosphere cell populations for the expression of CAR, the principal cell surface receptor for the human adenovirus [14], and its co-expression with Nestin and CD133. We detected Nestin expression on 90% of the neurosphere cells and CD133 on 15%–30% of the neurosphere cells. Of the CD133+ cells, 82% ± 3.1% expressed CAR, which is significantly higher (P = 0.02) than the CD133− neurosphere population (69% ± 4%) (Fig. 1A, 1C). Overall, CAR was expressed on ∼70% of all neurosphere cells. When we infected neurosphere cells with AdV-GFP and examined GFP expression in CD133 expressing cells versus the rest of the neurosphere population by flow cytometry, we detected a much higher percentage of CD133+ neurosphere cells (69% ± 13%) expressing GFP when compared to CD133− cells (51% ± 12%). The data were statistically significant based on the analysis of four independent experiments (two-tailed t-test, P < 0.05) (Fig. 1B, 1D).

Susceptibility of CD133+ and CD133− neural stem cells within neurospheres to AdV infection. (
Infection of neurosphere cells can be improved by dissociation
We investigated the infection efficiency of whole neurospheres versus dissociated neurospheres. First, we established a standard curve for estimating the cell number in a neurosphere of a particular size in order to establish the correct viral concentration for infecting whole neurospheres. We measured the diameter of 15 differently sized neurospheres and counted the number of cells in each sphere. From these data a formula for cell number (CN) as a function of the diameter (d mm) was generated CN(d) = 0.037 − (1 × 105 d) + 3.6 × 105 d 2 (r 2 = 0.9). The formula for the linear relationship between neurosphere volume (V = 4/3 πr 3) and cell number (CN(v) = 4.9 × 105 V) was computed based on the assumption that neurospheres are perfect spheres (Fig. 2A). This approximation proved reliable for estimating the cell number in a neurosphere based on the sphere diameter.
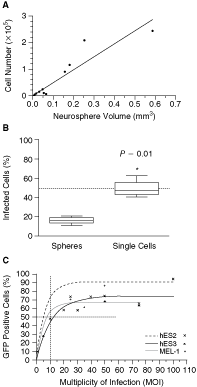
Infection efficiency of whole or dissociated neurospheres. (
To infect whole spheres with MOI 10, the number of cells in the spheres was determined using the approximation CN(v). For comparison, neurospheres were dissociated into a single cell suspension, counted by conventional cell count, and infected at MOI 10. Flow cytometric analysis 3 days after infection revealed 17% ± 2% GFP-positive cells in the infected whole neurospheres and 50% ± 5% GFP-positive cells in the neurospheres dissociated prior to infection. The 2.9-fold increase in GFP expression by dissociation reached significance (P = 0.01) by t-test (Fig. 2B), and was the method chosen for all the following experiments.
GFP expression in neurosphere cells over time and with increasing MOI
For optimal infection it is important to maximize infection and minimize viral load. We infected dissociated hES2, hES3, and MEL-1 neurospheres with MOIs ranging from 0.1 to 100 and examined the GFP expression 3 days after infection by fluorescence microscopy. GFP expression was clearly visible in some cells when infected at MOI 1, and the majority of cells expressed GFP when exposed to MOI 25 (Fig. 3A–3C). We also quantitated the number of cells expressing GFP in all three hESC-NSC lines by flow cytometry. Best-fit analysis revealed a nonlinear relationship between infection efficiency (I E) and the MOI defined by the maximal infection (I max) and the MOI needed to achieve the half-maximal infection (MOI0.5). All three neurosphere lines showed similar infection curves reaching similar maximal infection (Fig. 2C). Notably, infection efficiency reached a plateau when using MOI > 25 for all three lines, and increased viral load did not improve infection efficiency. Further, higher MOI increasingly impacted on cell viability and the ability to reform spheres (data not shown).
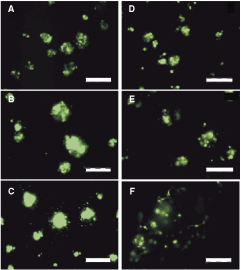
Evaluation of green fluorescent protein expression. (
GFP expression was followed up over 6 weeks by microscopy and images were taken each week using fixed exposure settings. Infected cells started to express GFP within 24 h and strong expression was detectable from day 3 of infection up to day 14 (Fig. 3D–3F). The intensity of GFP expression as well as the number of positive cells decreases thereafter. However, in some cells GFP expression was still visible up to 6 weeks after infection.
Virus infection does not change Nestin and CD133 expression in neurospheres
Neurospheres derived from hESCs have been shown to contain heterogeneous cell types, including NSCs and their progenitors [7], which may be important target populations for modeling different pathological events. In order to use neurospheres and AdV vectors for modeling such events, it will be important to assess the nonspecific viral effects on hNSC behavior. We infected neurospheres with AdV-GFP at various MOIs and assessed CD133 and Nestin expression by flow cytometry. There was no significant effect of viral infection on the percentage of Nestin-expressing cells in MEL-1 neurospheres up to MOI 25 (P > 0.05) (Fig. 4A). Similarly, the percentage of CD133+ cells in infected MEL-1 and hES2 neurospheres remained unchanged by viral infection (Fig. 4A), although there was a trend toward decreased marker expression at MOI ≥ 25.
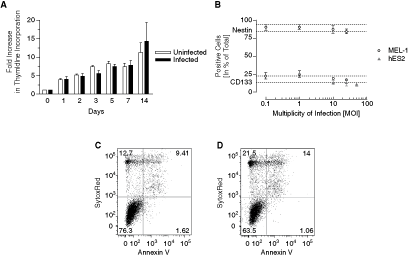
Cell growth and apoptosis after infection. (
Proliferation of neurosphere cells is not affected by AdV-GFP infection but more cells are apoptotic
In order to assess the effect of AdV-GFP on cell proliferation, dissociated neurospheres were infected with MOI 10 and proliferation was monitored over a period of 14 days using the thymidine-incorporation assay. First, there was no visible effect of the AdV-GFP on cell morphology and neurosphere formation. Second, even though there was no statistically significant difference between uninfected and infected cells throughout the 14-day period (P > 0.05), there was a trend toward slowed growth of the infected cells (Fig. 4B). To investigate the observed viral effect during the first 5 days further, we harvested cells at day 4 and measured AnnexinV and dead cells in the infected population and compared them by flow cytometry to the noninfected population. There was an increase in apoptotic and dead cells in the infected population when compared to uninfected cells (Fig. 4C, 4D), which is likely to be the cause of the observed slowed growth in the initial 5 days.
Infected neurospheres are multipotent
We tested infected hESC-derived neurospheres for their differentiation potential. Cells were plated onto laminin and cultured in neuronal differentiation medium for 14 days before they were examined for morphology and expression of the neuronal markers, βIII-tubulin and MAP2ab. Cells adhered over night and formed long projections within 7 days of culture (Fig. 5A). The cells co-expressed βIII-tubulin and MAP2ab (Fig. 5B). GFP was not co-expressed with either neuronal marker (Fig. 5C). Quantitative flow cytometry analysis of neuronal cultures revealed expression of βIII-tubulin in ∼80% of cells indicating that the majority of the cells were lineage committed (Fig. 5D). MAP2ab expression was detected on ∼17% of βIII-tubulin-expressing cells. Map2Aab was expressed without detectable βIII-tubulin in ∼15% of cells.
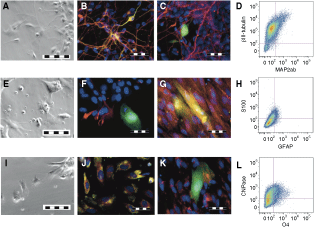
Neuronal and glial differentiation after infection. To assess neuronal differentiation, MEL-1 neurospheres were plated on laminin-coated coverslips and cultured for 14 days in NBM without EGF/FGF. (
Infected neurospheres were also differentiated on fibronectin under glial differentiation conditions for 14 days and examined using microscopy. Glial cultures contained a variety of cells with astrocyte and oligodendrocyte morphology (Fig. 5E, 5J). Some cells stained positive for GFAP and S100 confirming the presence of astrocytes (Fig. 5F). Very few of the astrocytes co-expressed GFP (Fig. 5G), indicating that the CMV promoter was repressed during glial differentiation. Quantitative analysis of astrocyte marker expression by flow cytometry analysis revealed that GFAP and S100 were co-expressed on a small subset of cells within the culture (∼5%) (Fig. 5H). We also assessed the glial cultures for oligodendrocyte marker expression and found a subpopulation co-expressing CNPase and O4 (Fig. 5K). However, these markers were also not co-expressed with GFP, indicating that cells with oligodendrocyte phenotype do not retain GFP expression throughout differentiation (Fig. 5L). Flow cytometric analysis of glial differentiation cultures indicated that CNPase was expressed on ∼65% cells and co-expression of CNPase and O4 in ∼16% of the cells (Fig. 5N).
We further quantitatively compared these results to assess potential differences in the proportion of neuronal and glial cells generated from infected versus uninfected hESC-derived neurospheres. As summarized in Table 1, we did not detect significant differences in expression of βIII-tubulin, GFAP, and O4 expression between infected and uninfected neurosphere cells (P > 0.05). We also confirmed the previous observation that more GFP expression is present in the glial cultures than in the neuronal cultures suggesting that expression is lost during neuronal differentiation but maintained in some glial cells (data not shown). In summary, infected neurospheres differentiated into neuronal and glial lineage cells in the same proportions as uninfected neurospheres.
D
All values defined as mean ± SEM of three experiments as determined by flow cytometry for aβIII-tubulin, bGFAP, and cO4.
Discussion
A more detailed understanding of stem cell biology, including the regulation of self-renewal and differentiation programs, will be required in order to use hNSCs to develop effective strategies to treat diseases such as cancer and neurodegenerative conditions. To address these issues, a variety of approaches to the generation, maintenance, expansion, and manipulation of stem cells in the laboratory are under investigation. In our laboratory, we used hNSCs derived from three different hESC lines after treatment with the BMP-2 antagonist, Noggin [8]. Using this technique, neurospheres were generated that exclusively contain neural cells including ∼15%–30% CD133+/Nestin+ hNSCs capable of differentiating into neurons, astrocytes, and oligodendrocytes [7].
Flow cytometry analysis of dissociated neurospheres generated from hESC lines hES2, hES3, and MEL-1 indicated that ∼70% cells express the adenovirus receptor, CAR, suggesting that most of these cells would be permissive to adenovirus infection. Indeed, at MOI 50 or greater, >70% dissociated neurosphere cells expressed GFP 4 days after infection. Adenovirus infection of dissociated neurospheres analyzed for co-expression of CD133 and GFP revealed a higher percentage of infected CD133+ NSCs compared to CD133− cells. The increased infection rates of CD133+ hNSCs correlated with the higher percentage of these cells expressing CAR and indicated that the CD133+ population is the most permissive cell type to adenoviral infection within the neurosphere population. Overall, these data indicate that CAR expression can be used as a valuable indicator for the susceptibility to adenovirus infection of neurosphere cells. A similar relationship between CAR expression and susceptibility to adenovirus infection has been reported in several other cell line models including embryonic stem cells (ESCs) [26], astrocytomas [27], and ovarian cancer cell lines [28].
Our results clearly showed that GFP expression was detected in more than twice the number of cells from dissociated compared to whole neurospheres using the same MOI. Not unexpectedly, this suggests that viral penetration of the neurospheres is limited and as the neurosphere diameter increases a higher percentage of cells are not exposed to the virus. Consequently, variability in sphere size significantly impacts on infection efficiency, which may at least partially explain inconsistencies in infection rates reported in other studies [17,20]. Using dissociated neurospheres we found that >60% of cells were infected at MOI 20 and that an increase in MOI above 25 did not increase infection rate greatly. Further, at MOI > 50 we observed a reduction in neurosphere reformation and proliferation, indicating significant virus-mediated side effects similar to those reported for rat fetal neurospheres infected at MOI 100 [18]. Additionally, although viral MOI 0.1–50 did not significantly alter CD133 or Nestin expression compared to uninfected cells, there was a trend toward decreased expression of these markers with increasing MOI. A decrease in Nestin expression has been described in murine NSCs 7 days after infection with Ad5 at MOI 20 [20]. Our data support the use of viral titers below MOI = 50 in order to minimize the viral effects for future analysis.
After infecting human NSCs, the AdV-GFP vector facilitates gene expression for up to 6 weeks. During that time, GFP expression declines with successive cell divisions, as the vector remains episomal and is not replicated [13]. However, GFP-positive cells remain clearly visible throughout this time. Similar observations have been reported for AdV infection of primary mouse NSCs [20]. Consequently, the adenovirus remains effective for at least 1 month and can be used for gene manipulation and subsequent experiments over this time course. In contrast, during the 14-day time course of the differentiation we observed a rapid decrease of GFP expression in glial cultures and an even more abrupt reduction in neuronal cultures. We could not detect GFP in cells that co-expressed neuronal markers and found only weak GFP expression in glial cells. This contrasts with observations in a rat model where CMV promoter-driven GFP expression is preserved during neuronal and glial differentiation [18,26]. Since hNSCs maintain GFP expression for up to 6 weeks, it is likely that the CMV promoter is repressed during differentiation.
At low MOI (<10), there was no significant difference in the proliferation of infected or uninfected cells measured over a 14-day time course. However, we did observe a trend toward slower proliferation of infected cells in the initial 3 days post-infection, which may be related to an increase in apoptosis detected during this time. Although at MOI 10 there was minimal effect on NSC proliferation, the initial increase in apoptosis would need to be taken into account when assessing the effect of AdV-mediated transgene expression in future studies. Variable effects of AdV infection on NSC proliferation have been reported in rodent and human models. For example, a significant reduction in the proliferation of rat fetal NSCs infected with AdV (MOI 100) compared to uninfected cells has been reported [18]. In contrast, no reduction in proliferation rates was detected in a study of human mesenchymal stem cell-derived NSCs infected with AdV-LacZ (MOI 100) [6].
In this study, we found that hESC-derived neurosphere cells could differentiate normally after being infected at MOI 10. Similarly, in rodent models, NSCs infected with higher viral concentrations were able to differentiate into neuronal and glial cells [18,20] although the latter study reported a decline in neuronal cells generated by AdV infected versus uninfected NSCs at MOI 20. We did not detect a significant difference between the percentage of infected and uninfected cells expressing neuronal or glial markers. However, we did observe a trend toward reduction in neuronal MAP2ab expression after infection, suggesting that the lower virus concentration may have lessened the impact on neuronal differentiation.
Combining our data and other published reports, it would appear that the effects of AdV infection on NSC function are variable and depend on the species, the manner in which the NSCs are derived, and the MOI used for experimental procedures. These findings suggest that careful optimization of AdV infection protocols is required to ensure the correct interpretation of the effects of virus-mediated transgene expression in NSCs obtained from different sources.
In summary, we have demonstrated that efficient AdV-mediated gene transfer into hESC-derived neurosphere cells can be achieved without significant effects on CD133 and Nestin expression, cell survival, cell proliferation rate, or differentiation potential. We anticipate that this model system will provide a practical new approach to study human NSC function in the context of neurodegenerative and neoplastic disorders.
Footnotes
Acknowledgments
The authors would like to thank Andrew Laslett and Pegah Jamshidi (Australia Stem Cell Centre, Clayton, Victoria, Australia) for providing us with MEL-1 neurospheres. Grant and funding support: NHMRC project grant 403982 (U.R.K. and P.B.D.), Australian Postgraduate Award, Jock and Marjorie Hetherington Award, and Stan and Jean Perron Award (C.M.B.), John Lillie Fellowship (P.B.D.).
Author Disclosure Statement
No competing financial interests exist.