
Abstract
The use of human stem cells (SCs) is a promising novel approach for the treatment of many diseases and injuries. Umbilical cord and amniotic membrane represent good sources for SCs, because they are abundant sources and there are less ethical issues unlike embryonic SCs. We aimed to isolate and characterize adult SCs from the subamnion region of the umbilical cord/amniotic membrane. Because mesenchymal stem cells (MSCs) are thought to show less immunogenicity, we first focused on the characterization of MSCs. Significant expression of typical SC-specific markers, such as SSEA-4, Oct-4, and Nanog was observed. Subamniotic MSCs did not lose the expression of Oct-4 and Nanog after freeze-thawing. Cell surface expression of MSC markers (CD73 and CD105) was confirmed by flow cytometry, and cells also differentiated into adipogenic, osteogenic, and chondrogenic lineages. On the other hand, typical embryonic SC-specific markers were not expressed and the cells also did not grow in soft agar. Thus, the subamniotic MSCs are distinct from embryonic SCs and do not show tumorigenicity in vitro. The cord lining membrane (subamniotic) MSCs isolated by our method maintain typical characteristics of MSCs in vitro, but also showed several specific features.
Introduction
T
Recently, several groups have reported the isolation of adult SCs or mesenchymal stromal cells from human amniotic fluid [9], amniotic membrane [10,11], and umbilical cord [12 –16]. Umbilical cord and amniotic membrane are attractive sources to obtain adult SCs due to total global abundances, ease of culture, and fewer ethical concerns unlike embryonic SC. Isolated SCs can still differentiate into many different lineages. Hence, the use of umbilical cord and amniotic membrane as sources of SCs could be one of the answers to the upcoming application of SCs in regenerative medicine.
In this study, we aimed to establish a protocol to isolate adult SCs from the cord lining membrane (subamniotic region of the umbilical cord), and characterize the isolated cells as a novel source for cell-based therapeutic approaches. We dissected the tissues without the use of proteases [17]. Isolated cells were mesenchymal (fibroblastic) cells. To our knowledge, this is a novel approach to the derivation, isolation, and characterization of MSCs. The cells satisfied the criteria of MSC, and significant expression of Oct-4, Nanog, and SSEA-4 was observed. We noticed that the expression pattern of SC markers is similar to epithelial SCs. Together with slight expression of cytokeratins, the cord lining membrane (subamniotic) MSCs (CL-MSCs) may be a progenitor of amniotic epithelium. Interestingly, we also detect CD14, which is normally expressed on monocytes and macrophages [18].
Materials and Methods
Isolation of MSCs from subamnion of human umbilical cord membrane
Human umbilical cord samples were obtained from healthy donors at the University of Texas Medical Branch hospital (Galveston, Texas) through the Tissue Bank of Shriners Hospitals for Children following approval of the institutional review board (IRB approval number #99-209, 03-247, 05-040). All tissues were tested for HIV, hepatitis, and mycoplasma. Briefly (See also Supplementary Fig. 1), (Supplementary materials are available online at http://www.liebertpub.com/) umbilical cord was washed well (3 times) with serum-free cold DMEM high-glucose medium containing 2× antimycotic (Invitrogen, Carlsbad, CA). Then the tissue was cut in pieces (∼1 inch long) and dissected to open the cord vessel. The pieces were put in 250-mm plastic petri dishes with medium for about 1 day in 5% CO2 incubator at 37°C. Wharton’s jelly absorbed DMEM (containing phenol red), therefore, it can be distinguished from the enveloping membrane of umbilical cord. The Wharton’s jelly was dissected using razor blades and pieces of outer envelope membranes were cultured after rinsing (Supplementary Fig. 1). CMRL1660 medium (Invitrogen) containing
Induction of osteogenic/adipogenic/chondrogenic differentiation
Both osteogenic and adipogenic differentiation were induced as reported by De Coppi et al. [9]. For osteogenic differentiation, cells were cultured with DMEM low-glucose containing 10% FBS (both from Invitrogen), penicillin/streptomycin, and osteogenic supplements [100 nM dexamethasone (Sigma, St Louis, MO), 10 mM β-glycerophosphate (Fruka, Steinheim, Germany), 0.05 mM ascorbic acid-2-phosphate (Fruka)]. For adipogenic differentiation, adipogenic supplements [1 mM dexamethasone, 1 mM 3-isobutyl-1-methylxanthine, 10 µg/mL insulin, 60 µM indomethacin (all from Sigma)] replaced osteogenic supplements. We also observed that significant decrease of number of cells during adipogenic differentiation, therefore, 3,000 cells/cm2 was not enough to obtain sufficient numbers of cells in our condition. Cells were directly plated onto a coverslip, and stained with Oil Red-O solution after 14 days. Oil Red-O staining was conducted as described in [19]. Chondrogenic differentiation was carried out as described in [20]. TGF-β3 (Peprotech, Rocky Hill, NJ) was added in the culture. Twenty-one days after the culture, the cell pellets were fixed with 10% formalin, embedded in paraffin and sliced into 5–10 µm sections. Safranin O was used to stain glycosaminoglycans [19].
Antibodies
Antibodies used in this studies were obtained from following sources; anti-SSEA-1 (clone MC-480), anti-SSEA-4 (clone MC-813-70), anti-TRA-1-60 (clone TRA-1-60), anti-TRA-1-81 (clone TRA-1-81), anti-TRA-2-54 (clone TRA-2-54/2J) were from Chemicon (Temecula, CA); anti-c-Kit (H-300), anti-Oct-3/4, (H-134) anti-Nanog (H-155) (all polyclonal) Abs, and anti-CD29 (β1 integrin) mAb (clone 4B7R) from Santa Cruz Biotechnology (Santa Cruz, CA); phycoerythrin (PE)-conjugated antiCD73 mAb (clone AD2) from BD Pharmingen (San Jose, CA); PE-conjugated anti-CD105 (clone SN6), anti-CD146 (clone P1H12), and anti-HLA-A/B/C (clone W6/32) from eBioscience (San Diego, CA); anti-CD10 (clone MEM-78), anti-CD13 (clone WM15), anti-CD14 (clone HCD14), anti-CD23 (clone D3.6), anti-CD31 (clone WM59), anti-Thy1 (CD90) (clone 5E10) from BioLegend (San Diego, CA); anti-CD34 (clone QBEnd-10) from Dako (Glostrup, Denmark); anti-MHC class II and isotype controls from Abcam (Cambridge, MA); anti-CD45 from BD Biosciences (San Jose, CA); anti-α-smooth muscle actin (clone 1A4), anti-desmin (clone DE-U-10), anti-pan-cytokeratin (clone C-11), and anti-vimentin (clone V9) were purchased from Sigma. Anti-STRO-1 [21] was a kind gift from Dr. Torok-Storb (Fred Hutchinson Cancer Research Center, Seattle, Washington), and anti-CD44 mAbs (clone A3D8 [22] and clone Hermes-1 [23]) were kindly provided by Dr. Haynes (Duke University, Durham, North Carolina) and Dr. Butcher (Stanford University, Palo Alto, California). All dye-conjugated secondary antibodies were purchased from Jackson Immunoresearch Laboratories (West Grove, PA).
Immunofluorescent staining
MSCs were trypsinized and seeded onto coverslips coated with 200 µg/mL of rat tail collagen I (Sigma-Aldrich). Cells were cultured in a CO2 incubator for 2 days and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. If permeabilization is required, 4% paraformaldehyde solution was discarded and then 0.1% Triton X-100 (w/v) in PBS was added and kept incubated for 5 min. Remaining aldehyde was quenched by sodium borohydrate. After rinsing with PBS, nonspecific binding was blocked by incubating 3% skim milk in PBS for 1 h. Each primary Ab was diluted in the blocking buffer and incubated with samples for 1 h at room temperature. Dilution of each primary Abs; anti-SSEA-1, 1/100; anti-SSEA-4, 1/200; anti-TRA-1-60, 1/100; anti-TRA-1-81, 1/100, anti-TRA-2-54, 1/100; anti-c-Kit, 1/500; anti-Oct-3/4, 1/100; anti-Nanog: 1/100; anti-CD34 and CD45, 1/100. After washing 3 times (5 min for each wash) with PBS, then samples were incubated with PBS containing X-rhodamine-conjugated donkey antimouse IgG, antimouse IgM, or antirabbit IgG secondary antibody. To image cytoskeletal components, trypsinized cells were seeded onto coverslips without coating. After 2 days, cells were fixed with 0.5% formaldehyde/0.5% glutaraldehyde/0.5% Triton X-100 (w/v) in PHEM buffer at room temperature for 5 min following post-fixation with 1% formaldehyde in PHEM buffer at room temperature for 10 min [24]. After quenching the remaining aldehyde with sodium borohydrate, nonspecific bindings were blocked with 5% (v/v) boiled donkey serum in PHEM buffer for 1 h at room temperature, then incubated with an appropriate primary antibody and a secondary antibody (Dilution of each primary Abs; anti-vimentin, 1/400; anti-α-smooth muscle actin, 1/1,000; anti-desmin, 1/40; anti-pan-cytokeratin, 1/100).
Microscopy and quantification
Phase contrast images (Fig. 1) were acquired by Diaphot 300 (Nikon, Japan) equipped with CoolSnap ES2 cooled charged devices (CCD) camera (Roper Scientific, AZ) and Plan DL 10× Ph1 objective lens (N.A. = 0.3). For immunofluorescent staining, both fluorescence images and phase contrast images were acquired by TE-800 (Nikon) equipped with CoolSnap fx CCD camera (Roper Scientific) and Plan Fluor 20× ELWD Ph1 DDL objective lens (N.A. = 0.5). For histochemical staining, DXM1200F color CCD camera (Nikon) was attached to the second port of the same microscope. Images were acquired with MetaMorph software (Molecular Devices, Downingtown, PA). To quantitatively analyze the results of immunofluorescent staining, we set a threshold for each image based on a negative control to create binary images, which were then overlaid with the corresponding phase contrast images to identify the position of cells in the field. Cells with binary signal “1” were counted as positive (Supplementary Fig. 2).
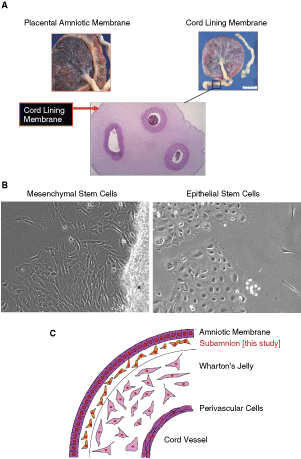
An overview of the isolation of cells. (
Flow cytometry
One 100-mm dish was used for each analysis. Adherent cells were harvested with mild trypsin treatment (0.05%). After quenching trypsin with serum containing media, cell pellets were washed once with PBS and incubated with 10% human serum (Jackson Immunoresearch Laboratories) in DPBS for 1 h at 4°C to block the binding of Fc region of antibodies to potential cell surface IgG receptors [25]. Then cells were incubated with 1 µg of appropriate primary antibody or isotype control antibody in 100 µL of PBS for 1 h at 4°C. After washing 3 times, pellets were resuspended in the same volume of PBS containing 1 µg of Cy2-conjugated donkey antimouse secondary antibody. Cells were washed 3 times, similar to previous washing, and then fixed with 1% paraformaldehyde in PBS for 20 min. All samples were analyzed by a flow cytometer FACSan (Becton Dickinson, Franklin Lakes, NJ) equipped with 15 mW 488 nm argon laser. All data were analyzed by Win MDI 2.9 (obtained from The Scripps Research Institute, La Jolla, CA).
RT-PCR
Three 60-mm dishes were used for each sample. Total RNA was extracted using Qiazol (Qiagen, Valencia, CA) according to manufacture’s protocol. RNA pellet was dissolved in RNase-free water and the concentration was calculated from absorbance at 260 nm. Each total RNA (10 µg) was treated with DNase I (RNase-free, New England Biolabs, Ipswich, MA) at 37°C for 30 min to get rid of contaminated genomic DNA. RT reaction was performed with random primer and M-MLV reverse transcriptase (Promega, Madison, WI) at 37°C for 30 min. Each specific PCR product was amplified by iCycler (Bio-Rad, Hercules, CA) using the primer sets (final concentration; 250 nM) shown below in a final reaction volume of 25 µL including 0.625 U of Go Taq DNA polymerase (Promega). The templates were heated at 95°C for 2 min, then amplified to appropriate cycles of 95°C (30 s), appropriate annealing temperature (30 s), 72°C (1 min) followed with one cycle of final extension at 72°C for 10 min. Primer sequences and PCR conditions (cycle number, annealing temperature, product size) for Oct-4, Sox-2, Rex-1, FGF-4, teromerase reverse transcription (TERT), Nanog, and GAPDH are as follows (Oct-4, Sox-2, Rex-1, FGF-4, TERT [10]; Nanog and GAPDH; [26]). Oct-4 forward, 5′-GAGGAGTCCC AGGACATGAA-3′; Oct-4 reverse, 5′-GTGGTCTGGCT GAACACCTT-3′ (45 cycles, 57°C, 151 bp): Sox-2 forward, 5′-GCCGAGTGGAAACTTTTGTC-3′; Sox-2 reverse, 5′-GTTCATGT GCGCGTAACTGT-3′ (33 cycles, 57°C, 264 bp): Rex-1 forward, 5′-GCGTACGCAAATTAAAGTCCAGA-3′; Rex-1 reverse, 5′-CAGCATCCTAAACAGCTCGCAGAAT-3′ (45 cycles, 57°C, 306 bp): FGF-4 forward, 5′-CTACAACGCCT ACGAGTCCTACA-3′; FGF-4 reverse, 5′-GTTGCACCAGAA AAGTCAGAGTTG-3′ (45 cycles, 57°C, 370 bp): TERT forward, 5′-AGAGTGTCTGGAGCAAGTTGC-3′; TERT reverse, 5′-CGTAGTCCATGTTCACAATCG-3′ (45 cycles, 57°C, 185 bp): Nanog forward, 5′-AGAAGGCCTCAGCACCTAC-3′; Nanog reverse, 5′-GGCCTGATTGTTCCAGGATT-3′ (35 cycles, 51°C, 205 bp): GAPDH forward, 5′-GTCAGTGGTGG ACCTGACCT-3′; GAPDH reverse, 5′-CACCACCCTG TTGCTGTAGC-3′ (35 cycles, 57°C, 255 bp). PCR products were analyzed by 2% agarose gels stained with ethidium bromide.
Colony-forming assay
Colony-forming assays were performed as described in [27]. MSCs were plated on 35-mm plastic petri dishes at 10 cells/cm2 followed by 14 days of culture. Then medium was removed and cells were stained with 0.5% Crystal Violet (Sigma) in methanol for 5 min. The dishes were washed twice with distilled water and dried before counting.
Soft agar assay
Soft agar assays were performed as described in [28]. 105 MSCs or NCCIT (human teratoma cell line as a positive control) were trypsinized and suspended in media (CMRL 1066 for MSCs and RPMI1640 for NCCIT) containing 20% serum and 0.3% Noble agar (Difco, Sparks, MD). The cell suspension was placed in a 6-well plate above a layer of solidified 0.6% Noble agar in media containing 20% serum. After the upper layer (containing cell suspension) was solidified, then 2 mL of media was added to each well. The media was replaced with fresh one every 5 days, and presence of colonies (>32 cells; 5 doublings) was determined under phase contrast microscopy after 7 and 14 days.
Results
Isolation of CL-MSCs
Figure 1A depicts the sources used in this study to obtain CL-MSCs. Freshly isolated tissues were cut and cultured as described in Materials and Methods. Approximately 10 to 14 days after starting the culture, a significant number of cells migrated from the implants into the petri dishes. Morphologically, most of cells appeared to be fibroblastoid (Fig. 1B, left), but we could also see a small population of epithelial-like cells when amniotic membrane was used as a source (Fig. 1B, right). To focus on fibroblastic cells, we used the subamnion region of the umbilical cord (cord lining membrane) as a source to obtain cells in this study (Fig. 1C). Supplementary Figure 1 shows the procedure, and isolated cells were confirmed to be mycoplasma-free (Supplementary Fig. 3).
Characterization of cell surface antigens
First, quantitative immunofluorescent microscopy (Supplementary Fig. 2) was used to measure the expression of several SC marker molecules. Figure 2A shows the representative images of immunostaining with anti-SSEA-4, anti-c-Kit, anti-Nanog, and anti-Oct-3/4. Many cells were positively stained with anti-SSEA-4 mAb. Permeabilized cells were also stained with anti-Oct-3/4 and anti-Nanog Abs, therefore expressing typical SC markers (Fig. 2A). Although immunostaining showed weak signals (Fig. 2A), we detected a significant expression of c-Kit (stem cell factor receptor) by immunoblotting (Fig. 2A inset). Figure 2B shows quantified results of several SC-related antigens. It should be noteworthy that 100% of cells expressed Nanog, which is one of the key molecules necessary for the maintenance of self renewal of SCs [29]. The other anti-Nanog Ab also showed that all cells in the field of view were Nanog positive (data not shown).
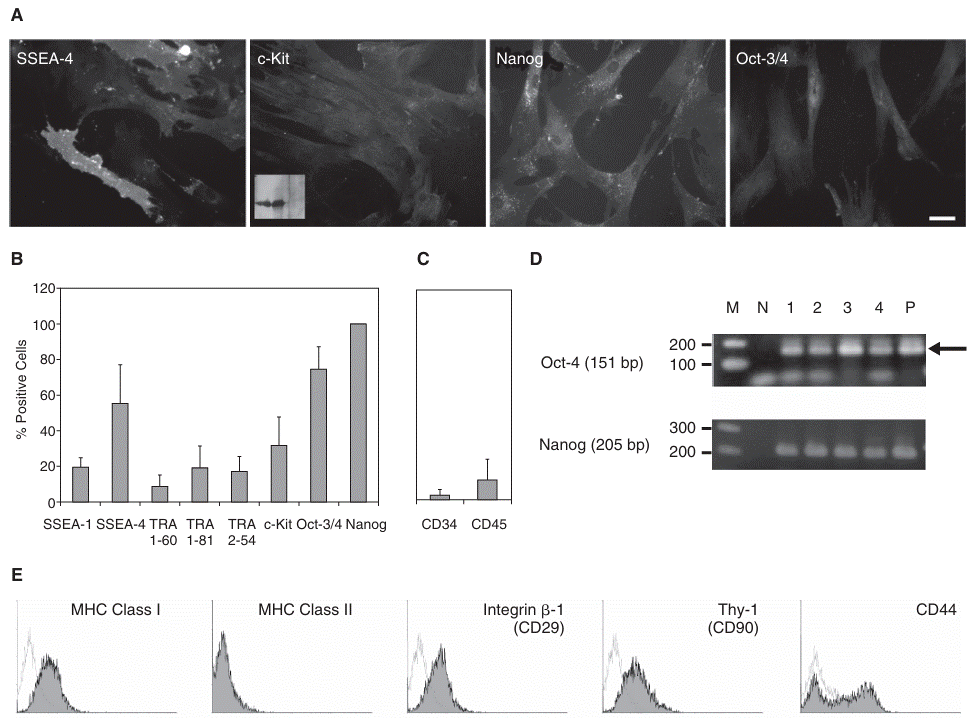
Analyses of stem cell markers expressed in the mesenchymal cells. (
To confirm that the cells are not derived from hematopoietic cells, we checked the expression of CD34 and CD45 on cell surface. We did not find significant CD34 and CD45 expression (Fig. 2C), indicating that the cells were not from hematopoietic origin. Although a part of cells apparently scored as positives, fluorescence signal of all CD34 and CD45 staining was nearly background level. Thus, we think that small positive fraction may not mean significant CD34/45 expression.
It is essential to know whether isolated adult SCs can be stored like regular cultured cells, and could be a key for expanding isolated SCs for large scale application in the future. It is, therefore, very important to know whether SCs can be frozen to stock. To determine if MSCs do not lose their capacity, we conducted RT-PCR analysis to measure the expression level of two typical master regulators maintaining self-renewal activity of SCs (Fig. 2D). The result indicates that freeze-thawing does not affect SC characteristics.
To check the expression of typical cell surface markers, we analyzed the expression of cell surface antigens by flow cytometry. As shown in Figure 2E, the cells expressed MHC class I, but did not express detectable amount of MHC class II, concurring with the previous report of the cells from the same tissue [9,14,15,30,31]. The cells also strongly expressed integrin β1 (CD29), Thy-1 (CD90), and CD44.
Expression of MSC markers on cell surface
To determine if our SCs meet the qualifying criteria of MSCs, we checked cell surface expression of CD73 and CD105, both of which are defined as MSC markers. Flow cytometry clearly showed that the isolated cells strongly expressed both CD73 and CD105 on the cell surface (Fig. 3A).
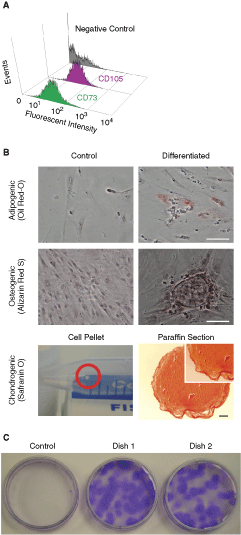
Characteristics of mesenchymal stem cells. (
Differentiation of CL-MSC
Another general defining criteria of MSC is the ability to differentiate into at least 3 lineages [32]. We examined if CL-MSCs can differentiate into these 3 lineages. Two weeks after differentiation, we could observe mineralization (osteogenic) or oil droplet (adipogenic) in some cells (Fig. 3B). After the chondrogenic differentiation, we observed the formation of rigid cell pellets (Fig. 3B). Control culture failed to form rigid pellets. Pellets were stained with Safranin O, suggesting the accumulation of glycosaminoglycan by chondrogenic differentiation (Fig. 3B).
CFU-F assay
To examine the growing capacity of CL-MSCs, we conducted CFU-F assay. As shown in the picture (Fig. 3C), CL-MSCs raised from a frozen stock grew well and formed numerous numbers of colonies (∼30 colonies), and diameter was clearly larger than 2 mm (Fig. 3C). This result indicates that isolated CL-MSCs retain the capacity to propagate. Relatively large colony size also suggests that CL-MSCs may actively migrate during proliferation, which is commonly observed in normal fibroblastic cells.
CL-MSCs are distinct from embryonic SCs
There is a consensus agreement that human embryonic stem cell (hESC) lines express Oct-4, Sox-2, FGF-4, teromerase reverse transcription (TERT), and Rex-1 [33]. Expression of these molecules in adult SCs seems to be controversial. Although Oct-4 is generally accepted as a SC marker, latter 4 molecules do not seem to be necessary to define adult SCs. We conducted RT-PCR with human teratoma cell line, NCCIT, as a positive control. Although Oct-4 was expressed as already shown in Figure 2D, CL-MSCs expressed neither Sox-2, FGF-4, TERT, nor Rex-1 (Fig. 4A). Thus, the result shows that CL-MSCs described here are distinct from embryonic SCs.
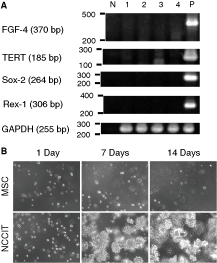
Embryonic stem cell marker expression and in vitro tumorigenic assay of mesenchymal stem cells. (
CL-MSCs did not show anchorage-independent growth
For future applications of CL-MSCs in regenerative medicine, it is important to know whether isolated CL-MSCs have tumorigenic potential or not. Anchorage-independent growth has been used for decades to simply measure tumorigenic activity of cells in vitro [34]. We plated CL-MSCs in a soft agar layer and observed colony formation. A Human teratoma cell line, NCCIT, was employed as a positive control. Figure 4B shows representative phase contrast images of each cell type in soft agar. After 2–3 days, CL-MSCs in soft agar layers start dying, and CL-MSCs were found not to form colonies during the 14 day period (Fig. 4B). In contrast, NCCIT cells grew up in soft agar layers 3 days after starting the culture, and already formed numerous numbers of colonies after 7 days (Fig. 4B). This result clearly indicates that CL-MSCs did not show tumor cell-specific features.
Origin of CL-MSC
To describe the specific feature of CL-MSCs, we tried to designate the origin and type of isolated CL-MSCs.
Because bone marrow-derived MSCs are well-studied, we first determined if the subamniotic MSCs share characteristics with bone marrow MSCs. STRO-1 [21] and CD23 [35] are known to be expressed in bone marrow–derived cells. Flow cytometric analyses showed that CL-MSCs expressed CD23 on cell surface, but not STRO-1 (Fig. 5).

Expression of bone marrow mesenchymal stem cells markers (CD23, STRO-1), endothelial markers (CD31), and other molecules (CD10, CD13, CD14, CD146). Dot line, isotype control; bold line, signal for each specific Ab.
Anatomically, the subamniotic region is adjacent to the outer envelop (amniotic membrane) of the umbilical cord [36]. Only the perivascular cells would be endothelial-like cells. We would like to ensure that CL-MSCs do not contain endothelial lineages. As shown in Figure 5, CL-MSCs did not express CD31. Interestingly, the cells expressed CD146 on the cell surface. Although CD146 was thought to be an endothelial marker, recent studies have shown that MSCs also express CD146 (see Discussion) [37,38]. Thus, the result suggests that endothelial or perivascular cells would not be contaminated in the isolated MSCs.
We also checked further molecules that more recently have been included in the criteria of MSC [32]. As similar to human umbilical cord matrix SCs reported by Weiss et al. [14], CL-MSCs strongly expressed CD10 and CD13 (Fig. 5). Interestingly, CL-MSCs also expressed CD14 (see Discussion) [18].
Because several cytoskeletal components, such as α-smooth muscle actin and intermediate filaments (vimentin, cytokeratin, and desmin), are considered good hallmarks to describe cell types and origin of the cells in tissues, we checked the expression of these cytoskeletal components in CL-MSCs by immunofluoresent microscopy. Several studies have shown that mesenchymal cells isolated from umbilical cord resemble myofibroblasts in terms of α-smooth muscle actin and vimentin expression [15,36,39]. Similar to umbilical cord and Wharton’s jelly mesenchymal stromal cells, CL-MSCs expressed significant α-smooth muscle actin and vimentin (Fig. 6). All the cells in the field were vimentin and α-smooth muscle actin positive. This result indicates that the CL-MSCs, reported here, share the similarity with myofibroblasts in terms of cytoskeletal composition, although we also noticed that the cells did not show well-developed fiber-like structure in α-smooth muscle actin staining [40] (Supplementary Fig. 4 shows immunostaining of α-smooth muscle actin in C2C12 myofibroblast). Instead, concentration of α- smooth muscle actin was often observed at the cell edges (Fig. 6). All the cells appeared to express vimentin, but interestingly, many cells did not show dense vimentin intermediate filament in lamella of cells, unlike the typical vimentin staining of fibroblasts [41]. On the other hand, it was demonstrated that immunohistochemistry of the whole umbilical cord showed heterogenecity of cytoskeletal components. As previously reported, basically the entire section of the umbilical cord expressed desmin [15,36]; however, umbilical cord–derived stroma cells lose desmin expression [15]. In accordance with a recent report, only trace desmin staining was observed in the cells (Fig. 6). The last component, cytokeratin, seems to be exclusively expressed in the amniotic epithelial layer and perivascular region of umbilical cord tissue [10,15,36]. Because CL-MSCs were isolated from the subamnion region, and expression pattern of SC markers resembled amniotic epithelial SCs (Fig. 2) [10], we determined whether CL-MSCs also expressed cytokeratins. Most cells did not show strong cytokeratin staining in contrast to epithelial SCs [10], although weak signal was observed in some cells (Fig. 6).
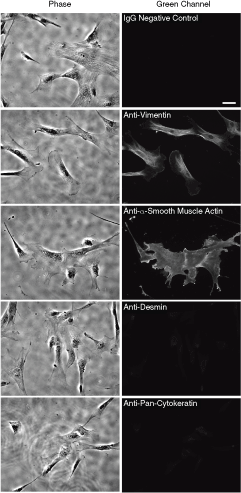
Cytoskeletal components in mesenchymal stem cells. Scale bar = 50 µm.
Discussion
In this study, we identified and characterized CL-MSCs from the subamnion region of the umbilical cord. Although Troyer and Weiss mentioned that 2 articles have been published showing the cells from subamnion [42], both articles actually described the cells from the amniotic fluid. Thus, at least in our knowledge, ours would be the first report showing the isolation and characterization of MSCs from subamnion (inner side of the cord lining membrane adjacent to amniotic epithelium). Numerous numbers of studies have reported multipotent stroma cells from the human umbilical cord [42]. However, the definition of MSCs is somewhat controversial, and the minimum criteria for MSCs needs <10 molecules and do not include any typical SC markers, such as Oct-3/4 and Nanog [32]. Thus, the nomenclature of many multipotent MSCs is suggested as multipotent mesenchymal stroma cells [32].CL-MSCs described here showed typical MSC characteristics, but additionally the cells also expressed a set of SC markers that the other reports from the same tissue did not [12,15,31]. The prominent feature of CL-MSCs is that the cells also express several SC markers in addition to MSC markers. Oct-4 and Nanog are well-accepted molecules to define SCs [43,44]. Our cells clearly expressed both molecules. We confirmed that many cells expressed SSEA-4, a stage-specific embryonic antigen mainly mark human embryonic SCs [45]. One recent study showed that SSEA-4 positive human bone marrow MSCs showed superior propagative activity [46]. Therefore, we suggest that CL-MSCs described in this study may retain a higher capacity than the cells reported from the same tissue.
Importantly, CL-MSCs did not express the other embryonic SC markers (Sox-2, FGF-4, TERT, and Rex-1), thus, the cells are still distinct from embryonic SCs. In addition, the cells did not show anchorage-independent growth in soft agar, which is a tumor cell-specific feature [34].
Miki et al. showed that epithelial SCs from amniotic membrane did not form tumors in nude mice [10]. Thus, although we cannot completely exclude the potential tumorigenicity in vivo, we think that CL-MSCs seem to be safe. Of course, we should carefully monitor that extracellular stimuli such as growth factors do not induce anchorage-independent growth of CL-MSCs in the future expansion of the culture system.
Unlike most reports from umbilical cord, CL-MSCs also expressed c-Kit. Mitchell et al. reported the isolation of c-Kit-expressing matrix cells from Wharton’s jelly that are capable of differentiating into neuronal cells expressing oligodendrocyte and astrocyte markers [47]. Although we did detect c-Kit by immunoblotting, immunostaining of cell surface c-Kit showed relatively weak signals. This observation probably suggests that majority of c-Kit may be retained in intracellular compartments. Flow cytometry also supported this idea (data not shown). It would be important to investigate if small amount of cell surface c-Kit is functionally significant or not. Nevertheless, c-Kit expression in CL-MSCs may suggest its high capacity to differentiate into several neuronal cell lines, which will expand the future applicability of CL-MSCs to treat neuronal diseases.
Fukuchi et al. reported that some batches of human placenta-derived MSCs cannot grow over 20 passages [48]. It is described that these cells were morphologically different (larger than proliferative cells). Karahuseyinoglu et al. also reported the similar phenotype recently, and suggested that cytokeratins are exclusively expressed in wide, flat cells, thought to represent loss of the proliferative population [15]. Although CL-MSCs slightly expressed cytokeratins, we assume that it is because of the similarity to epithelial SCs rather than the loss of proliferative activity. CFU-F assay indicated that CL-MSCs possess great capacity to expand.
Contamination of hematopoietic SCs is a concern when cells are isolated from tissues containing clotted human blood. Because CL-MSCs expressed c-Kit, which is expressed in hematopoietic progenitors, there may be some argument about the origin of the cells. But the cells are CD34 and CD45, markers for hematopoietic cells, negative. In addition, CL-MSCs strongly expressed CD105 (SH-2 antigen; endoglin/TGF-β receptor), which is not expressed in hematopoietic cells [49]. Surprisingly, we observed CD14 expression on CL-MSCs. Generally, CD14, known as a receptor for lipopolysaccharide and its binding protein [18], is thought to be expressed in monocytes and macrophages. It is possible that the cells might acquire CD14 expression after the isolation from a tissue, however, one study showed that CD14 is also expressed in human coronary artery smooth muscle cells [50]. Thus, CD14 may function as a sensor for MSCs to detect bacteria, which might explain how MSCs can home to injured/infected sites. Interestingly, soluble CD14 was reported to act a negative regulator of human T cell activation and function [51]. Although it has not been clear if cell surface CD14 can modulate T cell function yet, the significance of CD14 expression in CL-MSCs should be addressed in the future.
Bone marrow MSCs are the best characterized MSCs. We checked the expression of STRO-1 [21] and CD23 [35]. Although we did observe CD23 expression on cell surface, the cells did not express STRO-1. STRO-1 is sometimes used to enrich SCs from bone marrow [52], however, Prockop’s group reported STRO-1 negative cells retaining rapidly self-renewing SC subpopulation from bone marrow [19]. We found that some of the characteristics of CL-MSCs resemble their bone marrow MSCs in STRO-1, CD44, and c-Kit expression. Further investigation would be necessary to find if there is a clear marker to distinguish CL-MSCs from bone marrow MSCs.
During the dissection process of the umbilical cord tissue, we removed most parts from Wharton’s jelly, therefore, we think that fibroblastic cells shown here are derived from the subamnion region of the envelop membrane of umbilical cord. In fact, CL-MSCs looked to share same characteristics like amniotic epithelial SC [10]. Anatomically, subamnion locates adjacent to amniotic epithelium, and thus CL-MSCs may serve as an epithelial progenitor population. Trace amount of cytokeratin staining may also support this idea that CL-MSCs serve as a precursor of amniotic epithelial cells. Thus, we believe that there are several different populations of MSCs or multipotent cells in the umbilical cord tissue, and cells described here may be a progenitor of epithelial cells in umbilical cord envelop and amniotic membrane.
In conclusion, we successfully demonstrated the isolation and characterization of the subamniotic MSCs. Expression of many SC markers suggests their superior potential for future application and expansion. Some similarity of the cells to epithelial SCs implies that CL-MSCs may serve as an epithelial progenitor. CL-MSCs did not show tumor cell-specific growth in vitro, thus, the cells may be an attractive and rich source of MSCs. Further studies are required to understand the origin and function of CL-MSCs in the umbilical cord and to further investigate its clinical applicability.
Footnotes
Acknowledgments
We thank Drs. Eugene Butcher (Stanford University), Barton F. Haynes (Duke University), and Beverly Torok-Storb (Fred Hutchinson Cancer Research Center) for kindly providing us reagent, and Drs. Mark Furth and Anthony Atala (Wake Forest University) for providing us helpful tips. We also acknowledge Kimberly H. Palkowetz (Children’s Hospital, Department of Pediatrics, University of Texas Medical Branch (UTMB)) for flow cytometric analyses, Stephen Williamson (Tissue bank, Shriners Hospitals for Children) for collecting the tissues, and Drs. Massoud Motamedi (Center for Biomedical Engineering, UTMB) and Henry Epstein (Department of Neuroscience and Cell Biology, UTMB) for allowing us to use the optical imaging core facility. The authors also thank all administrative and lab personnel for their help, particularly Drs. Ahmed M. Al-mousawi and Felicia N. Williams for their useful comments, and Dr. Celeste C. Finnerty for showing a helpful website to search information regarding CD14. We also deeply appreciate the encouragement from all the local and scientific communities since after the landfall of hurricane Ike in Galveston. The authors apologize if any original articles were not cited in the article due to the space limitation. This work was supported by Anderson Foundation and Clayton Foundation for Research.
Author Disclosure Statement
T.T.P. is the founder and a shareholder of CellResearch Corp Pte Ltd and CordLabs Pte Ltd.