
Abstract
It has been demonstrated that human adipose tissue-derived mesenchymal stem cells (hASCs) enhance vascular density in ischemic tissues, suggesting that they can differentiate into vascular cells or release angiogenic factors that may stimulate neoangiogenesis. Moreover, there is evidence that shear stress (SS) may activate proliferation and differentiation of embryonic and endothelial precursor stem cells into endothelial cells (ECs). In this work, we investigated the effect of laminar SS in promoting differentiation of hASCs into ECs. SS (10 dyn/cm2 up to 96 h), produced by a cone plate system, failed to induce EC markers (CD31, vWF, Flk-1) on hASC assayed by RT-PCR and flow cytometry. In contrast, there was a cumulative production of nitric oxide (determined by Griess Reaction) and vascular endothelial growth factor (VEGF; by ELISA) up to 96 h of SS stimulation ( in nmol/104 cells: static: 0.20 ± 0.03; SS: 1.78 ± 0.38, n = 6; VEGF in pg/104 cells: static: 191.31 ± v35.29; SS: 372.80 ± 46.74, n = 6, P < 0.05). Interestingly, the VEGF production was abrogated by 5 mM N(G)-
Introduction
V
Several reports suggest that adipose tissue-derived mesenchymal stem cells (hASCs) differentiate into endothelial cells (ECs) after injection on ischemic hind limb mice models, which could participate, at least in part, in the new vessel formation [8 –10]. Additionally, there is evidence that hASC acquires an endothelial phenotype after being chemically treated in vitro [11 –13]. Thus, the purpose of the present study was to test the hypothesis that laminar SS induces endothelial phenotype in hASCs. Our data provided no evidence that laminar SS of 10 dyn/cm2 can induce the expression of endothelial cell markers in hASC, but it did show that SS stimulates nitric oxide (NO)-dependent vascular endothelial growth factor (VEGF) production in hASCs.
Materials and Methods
Isolation, ex vivo expansion of hASC
Subcutaneous adipose tissue was obtained from 24- to 53-year-old patients undergoing liposuction procedures. This protocol was approved by the University of São Paulo Ethics Committee (Protocol #16688/06). Cells were isolated from adipose tissue as previously described [14]. In brief, harvested tissue was dissociated by digestion with collagenase IA and centrifuged. The pelleted cells were then recovered and plated onto 10-cm culture plate (NUNC, Rochester, NY). Plating and expansion medium consisted of Dulbeccos’s modified Eagle’s medium (DMEM) low glucose with 10% fetal bovine serum and penicillin/streptomycin antibiotics (Invitrogen Corporation, Carlsbad, CA). After 24–72 h, cultures were washed in phosphate-buffered saline (PBS) in order to remove remaining erythrocytes and other unattached cells. Cells were maintained at 37°C with 5% CO2 in tissue culture dishes or flasks (Becton Dickinson, Franklin Lakes, NJ) until they reached 80% of confluence (usually within 5–7 days). Once 80% confluence was reached (P = 0), cells were detached with 0.5% trypsin–EDTA (Cultilab, São Paulo, SP, Brazil) and either replated at 1 × 104 cells/cm2 or used for experiments. This cell culture is well characterized in our laboratory that maintains constant doubling time (94.08 ± 4.88 h, n = 3–4) and population doubling (PD) per passage (1.11 ± 0.06, n = 4–9) and is non-senescent (Danoviz et al., unpublished data) up to the 14th passage. Therefore, at passage 14 the hASCs has experienced 15.87 ± 1.13 cumulative population doublings (CPD, n = 4–9). In addition, we have demonstrated adipogenic and osteogenic differentiation of these cells [15].
Shear stress experiments
Shear stress experiments were performed using a cone plate viscometer as previously described [16,17]. Cells (1.5 × 106) were plated into 15-cm plates. On the following day, fresh medium was added and cells were stimulated at 10 dyn/cm2 SS for 24, 48, and 96 h. During the experiment, the system was maintained at 37°C in humidified air with 5% CO2. Non-sheared cells were used as static control. N(G)-
Flow cytometry analysis
The immunophenotype of cultured hASC was analyzed by flow cytometry using the flow cytometer FACSCalibur (Becton Dickinson, San Jose, CA). Cells were harvested after being washed twice with PBS. Aliquots of 1 × 106 cells were incubated for 15 min at room temperature with FITC- or PE-conjugated monoclonal antibodies and washed twice in PBS containing 2% fetal calf serum and 0.1% sodium azide. The following fluorochrome-conjugated antibodies were used: CD13, CD29, CD31, CD14, CD34, CD44, CD45RO, CD49E, CD51/61, CD73, CD90, CD106, HLA-ABC, HLA-DR (BD Biosciences, San Jose, CA), and AC133 (Miltenyi Biotec, Bergisch Gladbach, Germany). A total of 10,000 events were acquired on a FACSCalibur flow cytometer and Cell Quest software (BD) was used for further analysis.
RT-PCR
Total RNA was extracted by the single-step method using Trizol reagent (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. RNA samples were quantified by absorbance at 260 nm and 280 nm in order to determine its concentration and purity levels. Only samples with 260 nm/280 nm index higher than 1.8 were considered. cDNA synthesis from total RNA (4 µg) was produced by reverse transcription (RT) using the High Capacity cDNA Reverse Transcription Kit according to the manufacturer’s protocol (Applied Biosystems, San Jose, CA).
Reverse transcription polymerase chain reaction (RT-PCR) was performed using Taq polymerase under the following conditions: initial denaturation at 95°C for 10 min; 40 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 1 min and extension at 72°C for 30 s; with final extension at 72°C for 5 min. The primers CD31, FLK-1, vWF and GAPDH were designed using the online software program Primer 3 (Primer 3, Ver. 3, Whitehead Institute/MIT Center for Genome Research http://frodo.wi.mit.edu/). Primers sequences and expected products lengths are shown on Table 1.
P
Abbreviations: FLK-1, VEGF receptor-2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; vWF, von Willebrand factor.
Determination of nitric oxide production
The NO production was evaluated by the amount of nitrite accumulation in the media of sheared and static cells using the colorimetric Griess assay as previously described [18]. In brief, 100 µL of culture medium or nitrite standards (serial dilutions of NaNO2 in non-conditioned media; Sigma-Aldrich, St. Louis, MO) were mixed with 100 µL of Griess reagent, containing 50 µL of 1% sulfanilamide and 50 µL of 0.1% naphtylethylene-diamine-dihydrochloride (Sigma) in 2.5 M H3PO4. Absorbance was measured at 540 nm. For short term of SS stimulation, the NO content was quantified with a Sievers NOA TM 280 nitric oxide analyzer, following manufacturer’s instructions.
Determination of VEGF-A
Cell culture medium was collected after 1, 24, 48, and 96 h from both sheared and static plates. The VEGF-A was measured by ELISA assay according to manufacturer’s instructions (R&D Systems, Minneapolis, MN).
Western blotting
Cells were washed with PBS and lysed in 20 mM Tris–HCl pH 7.5, 2 mM EDTA, 10 mM EGTA, 1% Triton X-100, 200 mM NaCl, supplemented with cocktails of protease and phosphatase inhibitors I and II (1:300; Sigma-Aldrich). After 10 min on ice, samples were centrifuged at 10,000g for 10 min to remove cellular debris. The 20–40 µg of cell lysates were run on SDS-polyacrylamide gels and transferred to PVDF membranes (Millipore, Billerica, MA). The immunodetection was performed by using rabbit polyclonal antibodies anti-eNOS (final concentration: 0.4 µg/mL), anti-nNOS (1.0 µg/mL; Santacruz Biotechnology, Santa Cruz, CA), anti-iNOS (1.0 µg/mL), anti-p-eNOS (0.5 µg/mL; BD Biosciences, San Jose, CA), anti-GAPDH (0.25 µg/mL; Calbiochem, San Diego, CA) and anti-p-AKT (0.25 µg/mL), and anti-AKT (1.0 µg/mL; Millipore, Billerica, MA). The secondary antibody-conjugated horseradish peroxidase (HRP) was either anti-rabbit IgG or anti-mouse IgG (Invitrogen, Zymed Laboratories, San Francisco, CA). Immunodetection was determined using the enhanced chemiluminescence (ECL) method. Densitometry measurement was made by using ImageJ software [19].
Statistical analysis
All data from multiple experiments are expressed as mean ± SEM assuming normal distribution of frequencies and equal variances between groups. Statistical analyses were performed using paired t-test or analysis of variance (ANOVA) (one or two way). When significant differences were detected (in ANOVA), comparison was carried out by Bonferroni post hoc test (GraphPad v.4.00 for Microsoft Windows). P < 0.05 was considered statistically significant.
Results
Shear stress does not induce the expression of endothelial markers in hASC
Cells submitted to laminar SS (10 dyn/cm2) up to 96 h were analyzed by flow cytometry for mesenchymal cell surface markers [8,20] and endothelial cell markers CD31 and CD34. Shear stress failed to modify the expression of the mesenchymal cell surface marker studied (Fig. 1A). More specifically, neither endothelial cell markers (CD31 and CD34) were induced nor mesenchymal cell markers (CD29 and CD90) were modified in sheared hASC up to 96 h (Fig. 1B).

Shear stress (SS) did not modify mesenchymal or endothelial cells markers profile. (
To further evaluate endothelial cell markers, time-course expression of CD31, vWF and FLK-1 genes were examined by RT-PCR (Fig. 1C). Similarly, no induction of CD31 expression was observed in hASC stimulated by SS. In addition, vWF and Flk-1 gene expression were not induced up to 96 h of SS.
Shear stress induces nitric oxide production on hASC
Shear stress stimulation of endothelial cells is accompanied by induction of nitric oxide (NO) release [21,22]. Although hASC submitted to SS failed to express endothelial markers, NO release was evaluated. Interestingly, shear stress lead to increased NO release by hASC after 24, 48, and 96 h of stimulation, measured as rate of production (data not shown) or accumulation in the media, which was blocked by 5 mM or 1 mM
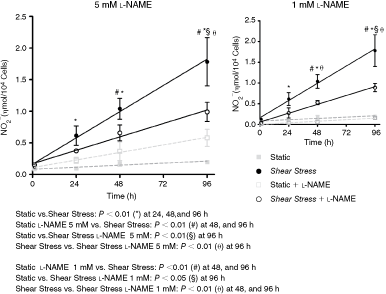
Shear stress (SS) induces nitric oxide production by human adipose tissue-derived mesenchymal stem cells (hASCs). Cells were subjected to shear stress for 24, 48, and 96 h and nitrite cumulative production was determined, with or without
We then examined NOS isoforms that could be mediating this NO production, hASC were assayed by western blotting after 96 h. Only endothelial nitric oxide synthase isoform (eNOS) was observed in both static and shear-stimulated conditions but we were unable to detect significant changes with this assay (Fig. 3A). In contrast, flow cytometry analysis showed an increase from 18.4% to 34.3% of hASCs expressing eNOS when hASCs were exposed to shear, consistent with the idea that eNOS is being stimulated under the present experimental conditions (Fig. 3B). Note that hASCs express lower levels of eNOS compared to endothelial cells (Fig. 3C).

Nitric oxide (NO) synthase determination in human adipose tissue-derived mesenchymal stem cells (hASCs). (
Shear stress induces VEGF production mediated by nitric oxide
NO induces VEGF release in endothelial, smooth muscle, and cancer lineage cells [23,24], so we then verified whether VEGF can be stimulated in ASCs exposed to SS. Shear stress induced VEGF accumulation in the cell culture medium and a constant increase on VEGF rate production similarly to NO release were observed (Fig. 4). Moreover, 5 mM or 1 mM
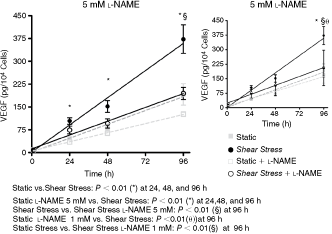
Effect of shear stress on VEGF production by human adipose tissue-derived mesenchymal stem cells (hASCs). Cells were subjected to shear stress for 24, 48, and 96 h and VEGF cumulative production was examined with or without
Discussion
In the current study, we addressed the issue of hASCs differentiation toward an endothelial cell phenotype by hemodynamic stimulus, particularly SS. Our data provide evidence that SS (10 dyn/cm2) for up to 96 h cannot induce the expression of specific markers for endothelial cells in hASCs, including FLK-1, vWF, and CD31, but it can increase the VEGF production by these cells. This is consistent with the lack of changes in cell morphology, that is, no cell alignment in the direction of flow (data not shown), which is typical for endothelial cells under shear condition [25]. Furthermore, there were no indications that the expression of mesenchymal cell markers decreased, suggesting that SS alone is not able to differentiate hASC into endothelial cells. This is in contrast with reports showing that SS increases the expression of endothelial markers in other cell types, including embryonic stem cells [26,27] and endothelial precursors [5]. It is important to emphasize, however, that there are evidences of hASCs differentiation into endothelial cells in vitro by chemical stimuli [11,12] and also in vivo [8 –10].
It is believed that the first commitment of embryonic stem cells to endothelial phenotype is genetically controlled. The fluid flow plays a role during the late stages of vascular formation participating in the maturation process of the vascular system [28]. Most of the studies that showed positive endothelial differentiation by SS used embryonic stem cells (most precursor cells) or endothelial precursors (endothelial pre-committed cells) ([29] for review). One may speculate that shear force alone is not sufficient to differentiate adult mesenchymal stem cells into endothelial cells. It appears that SS may be able to modulate cells that are already driven to express endothelial phenotype. In this context, Zhang et al. [30] demonstrated that biochemical stimulus and shear force act in synergism to endothelial differentiation of amniotic fluid-derived stem cells.
Additionally, during embryonic development, hemangioblasts (Flk-1+ cells) are able to lead both the hematopoietic and endothelial cells [31]. Hemangioblasts, when stimulated with cytokines, give rise to angioblasts, which also express FLK-1 and can migrate [32] and/or differentiate into endothelial cells to generate capillaries by VEGF signaling and other cytokines [33]. Zeng and colleagues showed that the cascade Flk-1-PI3K-Akt-HDAC3-p53-p21 is crucial in the differentiation process of ES into endothelial cell induced by SS or VEGF [26]. Unexpectedly, hASCs used in the present study do not express FLK-1. This observation seems to be exclusive for human cells since murine ASCs do express Flk-1 that is increased by SS (Nakamuta JS, unpublished data from our laboratory). The absence of Flk-1 in hASCs is consistent with their incapacity to differentiate into endothelial cells, since the presence of Flk-1 seems to be necessary for this process [10,34]. Thus, it is tempting to speculate that the modulatory role of shear stimulus may affect only cells expressing an endothelial phenotype (Flk-1+ cells).
Even though hASCs showed no evidence for SS-induced differentiation, an interesting observation was the increased NO production stimulated by SS, which is characteristic of endothelial cells. This response was partially inhibited by treatment of 5 mM
In addition to shear-induced NO production in hASCs, it was also observed an increase of VEGF release, which is dependent of NO. The VEGF is a key mediator of vascular permeability, angiogenesis, and inflammation processes intimately involved in tissue repair, leading to new blood vessels formation [41]. It is produced by a wide range of cells [42] including macrophages, vascular smooth muscle cells, pericytes, fibroblasts, keratinocytes, tumor cells, lymphocytes, megakaryocytes, neutrophils, basophils, mast cells, astrocytes, and bone marrow mesenchymal stem cells (MSC) [43]. hASC also produces VEGF that is increased under hypoxia [44] or treatment with cytokines [7], and in this work we demonstrated that SS induced VEGF release in hASCs (Fig. 4). The augmented VEGF mediated by NO is widely demonstrated in biological systems [45,46] and it is the first verification in hASCs submitted to SS. On the other hand, there is evidence of VEGF suppression by NO donor in MSCs. Wang et al. [43] demonstrated that NO donor, but not endogenous NO, can inhibit VEGF production. Evidences showing inhibition of VEGF release by NO are mostly observed by usage of NO donors [23,47], induction of endogenous NO usually results in augmented VEGF [48,49]. We believe that this opposite response is related to the amount of NO generated by these 2 stimuli [42,50] that can elicit distinct signaling pathways. The kinetic of NO release by NO donors are very distinct from cells stimulated to produce NO, and may result in different modulation of VEGF release [42,50].
Data from our laboratory have shown that local intramyocardial injection of ASCs alone and fibroblasts or myoblasts genetically modified to express VEGF in rat ischemic heart results in better cardiac performance (Nakamuta JS, et al. unpublished data, [51] and [52], respectively). Considering that ASCs showed basal production of VEGF that can be influenced by hypoxia or physical factors, as demonstrated in the present report, one may speculate that the benefit of ASC therapy in ischemic tissue is mediated, at least in part, by growth factors release including VEGF. Moreover, ASCs may also have a beneficial effect influencing other important processes such as inhibiting apoptosis [53], inflammation [54], and other immune responses [55], and be able to produce angiogenic factors under hypoxia [56,57].
Taken together, the data presented provide evidence that although SS failed to induce the expression of endothelial cell markers, it stimulated NO-mediated VEGF production, which may be a contributing factor to be explored for the use of these cells in ischemic tissue repair strategies.
Footnotes
Acknowledgments
V.B., V.G.B., and C.L.R. were recipients of training fellowships from Fundação de Amparo à Pesquisa do Estado de São Paulo—FAPESP—(2005/57591-4, 2006/52053-7) and Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq—(100041/2006-0), respectively. V.B. is enrolled in the Molecular Biology master program at the UNIFESP/EPM.
This work was funded by grants from FAPESP (07/58942-0, 08/52436-9, 08/52335-8) and CNPq (573887/2008-0). The authors want to thank Laura I. V. Brandizzi for technical assistance in measurements and Dr. Gustavo G. Duarte for assistance during the liposuction procedure.
Author Disclosure Statement
The authors declare no conflict of interest.