
Abstract
Human embryonic stem cells (hESC) can differentiate into a variety of specialized cell types, and they constitute a useful model system to study embryonic development in vitro. In order to fully utilize the potential of these cells, the mechanisms that regulate the developmental processes of specific lineage differentiation need to be better defined. The aim of this study was to explore the molecular program involved in the differentiation of hESC toward definitive endoderm (DE) and further into the hepatic lineage, and to compare that with primitive endoderm (PrE) differentiation. To that end, we applied two protocols: a specific DE differentiation protocol and an intrinsic differentiation protocol that mainly mediates PrE formation. We collected hESC, hESC-derived DE, DE-derived hepatocyte-progenitors (DE-Prog), DE-derived hepatocyte-like cells (DE-Hep), and the corresponding PrE derivatives. The samples were analyzed using microarrays, and we identified sets of genes that were exclusively up-regulated in DE derivatives (compared to PrE derivatives) at discrete developmental stages. We also investigated known protein interactions among the set of up-regulated genes in DE-Hep. The results demonstrate important differences between DE and PrE differentiation on the transcriptional level. In particular, our results identify a unique molecular program, exclusively activated during development of DE and the subsequent differentiation of DE toward the hepatic lineage. We identified key genes and pathways of potential importance for future efforts to improve hepatic differentiation from hESC. These results reveal new opportunities for rational design of specific interventions with the purpose of generating enriched populations of DE derivatives, including functional hepatocytes.
Introduction
H
Human ESCs spontaneously differentiate to heterogeneous populations of various cell types including extraembryonic PrE [9,10]. In addition, recent reports have shown successful differentiation of hESC to DE [1,2,8,11]. Some investigators reported on the further maturation of DE into beta cells [12] while others attempted to direct the cells toward the hepatic lineage [1,2,11]. Although the results are encouraging, the hepatocyte development was, in most studies, not efficient enough for applying the methods on a large scale. Moreover, despite improved protocols, the purity of the final cell preparation remains an issue, since the hepatic cell types are most often observed in heterogeneous cultures that also contain other cell types.
Interestingly, DE and PrE have been compared in the murine system [6,13], whereas similar studies in human cells are lacking. However, in a recent study conducted on hepatocyte-like cells derived from one hESC line, AFP-positive cells were identified, sorted, and selected for global transcriptional profiling [14]. It should be noted that AFP is expressed in both DE and PrE, as well as in other cell types, and not solely in hepatocytes [4,11,15]. Thus, monitoring expression of AFP alone is not sufficient to conclude a hepatic phenotype. Moreover, genetic variation between hESC lines is known to influence the differentiation properties, and additional studies employing more than one hESC line are needed [16 –18].
In the present study, we used two different protocols for the differentiation of hESC to hepatocyte-like cells: (1) a DE-protocol that efficiently directs hESC to DE and further toward the hepatic lineage [8,19] and (2) an intrinsic protocol (I-protocol) known to mainly mediate PrE differentiation [9,10] (Fig. 1). Using derivatives from these two protocols, we studied the temporal changes in gene expression during differentiation. For each time point, we compared the gene expression profile of cells derived through DE with cells derived through PrE. The experiments were repeated using three different hESC lines, and the well-characterized hepatocellular carcinoma cell line HepG2 was included as a reference. This experimental setup allowed the identification of sets of genes that were exclusively up-regulated during DE and PrE differentiation, respectively. The up-regulated genes were further classified into families according to functional annotation, followed by identification of protein interaction networks. Our results demonstrate important differences between DE and PrE differentiation on the transcriptional level, and we report a unique molecular program that is exclusively activated during development of DE and throughout differentiation toward the hepatic linage.

Experimental design. Human embryonic stem cells (hESC) (UD) were differentiated using two protocols: a definitive endoderm (DE) differentiation protocol (yellow) and an intrinsic protocol (blue) that mainly mediates primitive endoderm differentiation (PrE). The timeline represents the number of days in culture. The HepG2 cell line is included as a control. Three cell lines were included in the study: SA002, SA167, and SA461. For each cell line, the experiments were repeated twice. Color images available online at
Methods
Culture of hESC
The hESC lines SA002, SA167, and SA461 (Cellartis AB, Gothenburg, Sweden) were derived, cultured, and characterized as previously described [20]. In brief, hESC were propagated on mitomycin-C (Sigma-Aldrich, St. Louis, MO)-inactivated mouse embryonic fibroblast feeder layers in VitroHES medium (Vitrolife AB, Kungsbacka, Sweden) supplemented with 4 ng/mL human recombinant basic fibroblast growth factor (hrbFGF, here termed FGF2; Invitrogen/Gibco, Carlsbad, CA,
Differentiation of hESC using the DE-protocol
Differentiation of hESC into DE was performed essentially as described in Ref. [8] and further maturation into hepatocyte-like cells (DE-Hep) was carried out as described in Ref. [19]. In brief, before initiating differentiation into DE, the hESC cultures were washed with PBS+/+ (Invitrogen/Gibco) and thereafter cultured for 4 days in RPMI 1640 medium (Invitrogen/Gibco) supplemented with PEST (1%) (Invitrogen/Gibco), Glutamax (1%) (Invitrogen/Sigma), 100 ng/mL recombinant human Activin A, and 4 ng/mL FGF2 (both from R&D, Systems, Minneapolis, MN,
Subsequently, the medium was changed to Advanced RPMI 1640 medium supplemented with PEST (1%) (Invitrogen/Gibco), Glutamax (1%) (Invitrogen/Sigma), FBS (0.2%; Gibco), FGF1 (100 ng/mL; PromoKine, Heidelberg, Germany,
Differentiation of hESC using the I-protocol
The intrinsic differentiated hepatocyte-like cells were derived as previously described [9,10]. In this protocol, the hESC were differentiated for 20 days in VitroHES™ medium supplemented with 4 ng/mL FGF2. The medium was replaced with fresh medium after 10 days and again after ten additional days. The experiments were performed twice for each hESC line.
Culture of HepG2 cells
HepG2 cells (HB-8065, American Type Culture Collection, Manassas, VA,
Immunocytochemical methods and antibodies
Cells in culture were fixed in 4% (w/v) paraformaldehyde (PFA) for 15 min and then washed with phosphate buffer (PBS) twice. For visualizing extracellular proteins cells were blocked in 5% FBS in PBS for 30 min, for intracellular protein staining cells were also permeabilized in 0.1% Triton X-100 in PBS (0.1% PBT) for 30 min. Primary antibodies were diluted in 1% FBS in PBS and applied to samples and incubated overnight at 4°C. Primary antibodies used were mouse anti-CK18 (1:200; DAKO Cytomation), sheep anti-CYP3A4/7 (1:100; Biomol), rabbit anti-Sox17 (1:2,000, a kind gift from E. Baetge). To visualize the nucleus cells were incubated with DAPI at 0.05 mg/mL (Sigma) for 5 min at RT. Samples were washed with PBS twice before secondary antibodies were applied. The secondary antibodies were diluted in 1% FBS in PBS and incubated for 1 h at room temperature (RT). For the Cyp-staining, the cells were incubated with secondary antibodies in PBS for 3 h at RT. Secondary antibodies used were Cy3 donkey anti-mouse IgG (1:1,000; Jackson Immuno Research, Suffort, UK), Alexa Fluor 488 donkey anti-sheep IgG (1:1,000; Molecular Probes, Invitrogen/Gibco, Carlsbad, CA), Alexa Fluor 488 donkey anti-rabbit IgG (1:1,000; Molecular Probes). Dilution of the antibodies was made according to the manufacturer's recommendations. Samples were washed again with phosphate buffer twice and then mounted in DAKO Cytomation mounting medium prior to fluorescence microscopy analysis.
RNA extraction and microarray experiments
Total RNA was extracted from hESC and their derivatives at specific time points using RNeasy Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions. DNAse treatment was performed using an Rnase-free DNase Kit (Qiagen). The RNA quality and concentration was measured using an Agilent 2100 Bioanalyzer and Nanodrop ND-1000, respectively. Total RNA was processed following the GeneChip® Expression 3′-Amplification Reagents One-cycle cDNA synthesis kit instructions (Affymetrix Inc, Santa Clara, CA) to produce double-stranded cDNA. This was used as a template to generate biotin-targeted cRNA following the manufacturer's specifications. Fifteen micrograms of the biotin-labeled cRNA was fragmented to strands between 35 and 200 bases in length, 10 μg of which was hybridized at 45°C for 16 h to the HGU 133 Plus 2.0 genome array (Affymetrix, Santa Clara, CA) using standard procedures.
Data analysis
The data sets were normalized using the MAS5 software (Affymetrix, Santa Clara, CA) and probes with absent signals in all samples were excluded from further analysis.
Comparison of differentially expressed genes in samples derived from the DE- and I-protocols
Genes expressed differentially between the DE-protocol and I-protocol were identified by the following approach: (1) For each cell line and time point, probes that were up-or down-regulated with fold change (FC) >3 compared to hESC in each of the three cell lines were selected. (2) For each of the three time points (4, 10, and 20 days), lists were compiled summarizing: (i) probes that were differentially expressed in both protocols, (ii) probes that were differentially expressed in the DE-protocol but not in the I-protocol, and (iii) probes that were differentially expressed in the I-protocol but not in the DE-protocol. This part of the analysis was performed at the probe level and it should be noted that each gene is represented by one or several probes on the arrays. Consequently, probes representing transcripts from the same gene may in some cases be assigned to different groups.
Gene ontology analysis
To further investigate the biology of the up-regulated genes in DE-Hep, the DAVID tool [22] was used to explore their gene ontology annotations according to biological process molecular function and cellular component [23].
Analysis of up-regulated pathways in DE-Hep and PrE derivatives
To identify significantly up-regulated pathways during DE-Hep differentiation, the KEGG pathway database [24] was searched using the DAVID software tools [25]. By comparing with a reference list containing all genes in the human genome, significant overrepresentation of up-regulated genes in a particular pathway can be calculated. Pathways containing significantly (P < 0.05) more up-regulated genes than expected by chance were identified using a modified Fisher's exact test. For comparison, an identical analysis was conducted using the list of up-regulated genes (FC > 3) in the PrE derivatives generated by the I-protocol.
Protein interaction network analysis
To investigate possible interactions between the gene products from up-regulated genes in DE-Hep and PrE derivatives, the STRING search tool [26,27] was used for creation of protein interaction networks (PIN) as previously described [28]. One PIN was created from each of the lists of up-regulated genes in DE-Hep and PrE derivatives, respectively. These networks were explored and compared based on topological characteristics, and gene products (proteins) with high connectivity (hubs) were identified. In accordance with Han et al., we defined hubs as proteins with ≥5 interactions with other proteins [29].
Real-time quantitative PCR
Total RNA was extracted from hESC and their derivatives at selected time points for cell line SA002 and DNAse treated, all as described above. cDNA synthesis was performed using a High-Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer's protocol. TaqMan-based Assay on demand pre-designed primers and probes for the genes: POU5F1 (Oct4), CXCR4, SOX17, CER1, AFP, and ALB were used for PCR in a TaqMan ABI 7500 sequencer (Applied Biosystems). Relative quantification for a given gene, expressed as relative mRNA levels compared to the control (hESCs), was calculated after normalization to HPRT and using the ΔΔCT formula.
Results
On a morphological level, there is a clear difference between cells obtained from the DE- and I-protocols. The I-protocol generates heterogeneous cultures of cells with various morphologies (data not shown) while the DE-protocol generates much more homogeneous cultures with predominantly triangle-shaped endodermal cells (Fig. 2A). Immunocytochemical analyses further confirm the DE differentiation and underscore the difference between the 2 protocols. Known from present and previous results [19], the DE cells are positive for SOX17 (Fig. 2C) and CXCR4, and negative for SOX7 and OCT4 (POU5F1), whereas PrE cells are positive for SOX17, SOX7, OCT4 and negative for CXCR4. When the cells further mature toward the hepatic lineage (DE-Hep), the morphology of the cells gradually change and resemble human hepatocytes with more than one nucleus (indicated by arrows in Fig. 2B), polygonal shapes, and the cells are also starting to form substructures. The DE-Hep stain positively for CK18 (red), CYP3A (green), and DAPI (blue) staining show that many cells are binucleated (Fig. 2D).
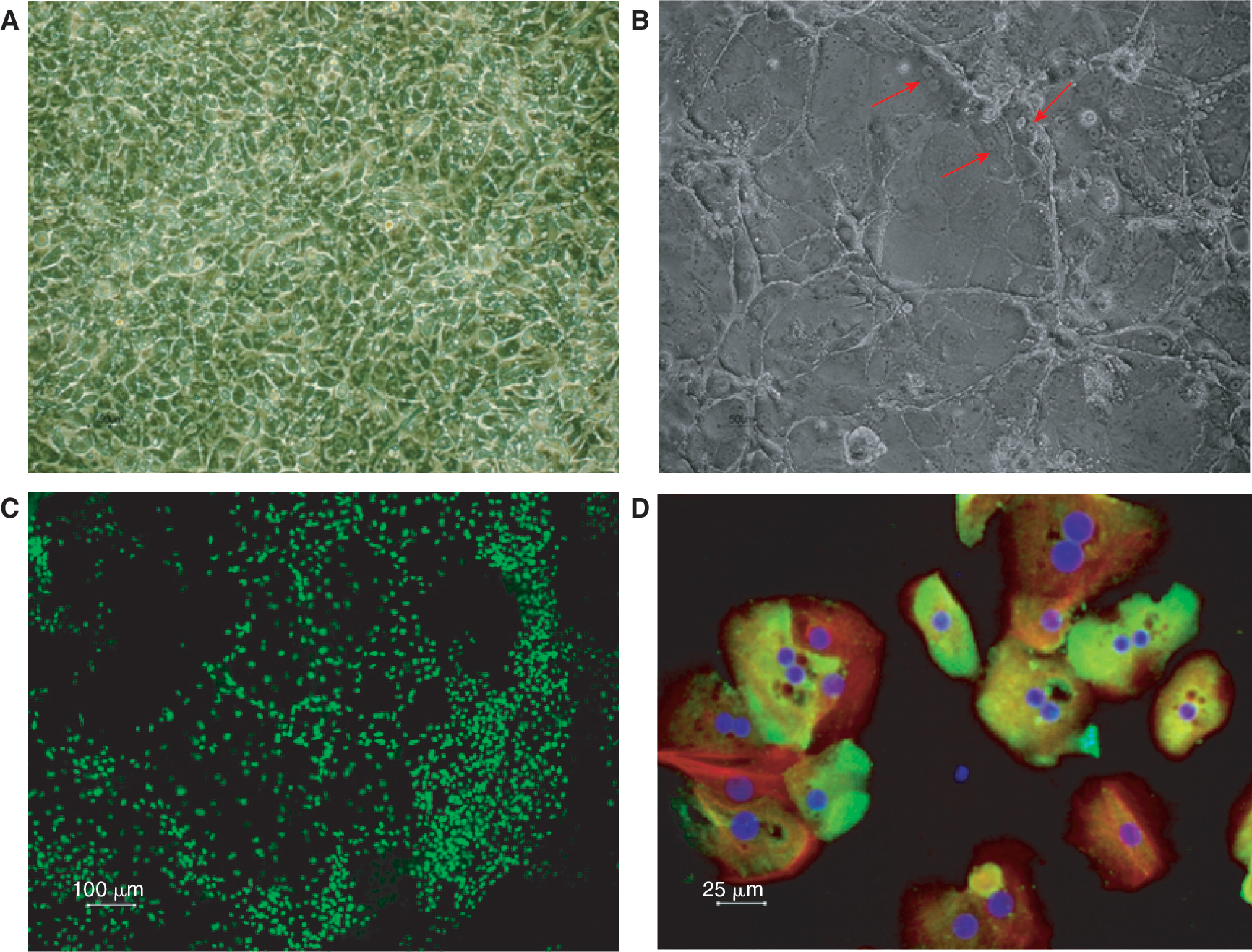
Morphology and immunocytochemistry of definitive endoderm (DE) and DE-derived hepatocyte-like cells (DE-Hep). Panels
Gene expression profiles of marker genes for hESC, DE, PrE, and derivatives thereof
To verify that the differentiation regimes applied in this study efficiently induced hESC differentiation, the RNA levels of known markers of pluripotency were monitored. Figure 3 shows the kinetics of the down-regulation of OCT4, SOX2, DNMT3B, and NANOG in cells cultured according to the DE- and I-protocols.

Marker genes for pluripotency. Blue bars represent cell line SA002, green bars represent cell line SA167, and orange bars represent cell line SA461. Left-side diagrams show expression levels when using the definitive endoderm (DE)-protocol (solid bars) and right-side diagrams show expression levels when using the I-protocol (striped bars). UD indicates undifferentiated human embryonic stem cells (hESC). The intensity units are directly proportional to the mRNA level. The figure shows a distinct down-regulation of these markers for pluripotency as the cells differentiate. Color images available online at
Furthermore, marker genes for specific developmental stages during endodermal differentiation were investigated and compared between the two differentiation regimes and the results are shown in Figures 4 –6. Increased mRNA levels for well-known markers for DE, such as CXCR4, GSC, CER1, SOX17, NODAL, HHEX, and FOXA2, were observed in DE at day 4 (Fig. 4A) [3,8,11,30,31]. In addition, AFP and HNF4A, which are known to be expressed at higher levels in the extraembryonic PrE than in DE during early differentiation [32], display an expected pattern (Fig. 4B).

Marker genes for early endoderm. Panel
A group of genes reported to be expressed during initiation of hepatocyte differentiation were also monitored [1,3,11,15,33,34]. Figure 5 shows the expression levels of AFP, DOK4, HAL, PROX1, TAT, and WNT2B. These genes show higher expression at day 10 in the samples differentiated according to the DE-protocol than in the corresponding PrE derivatives.
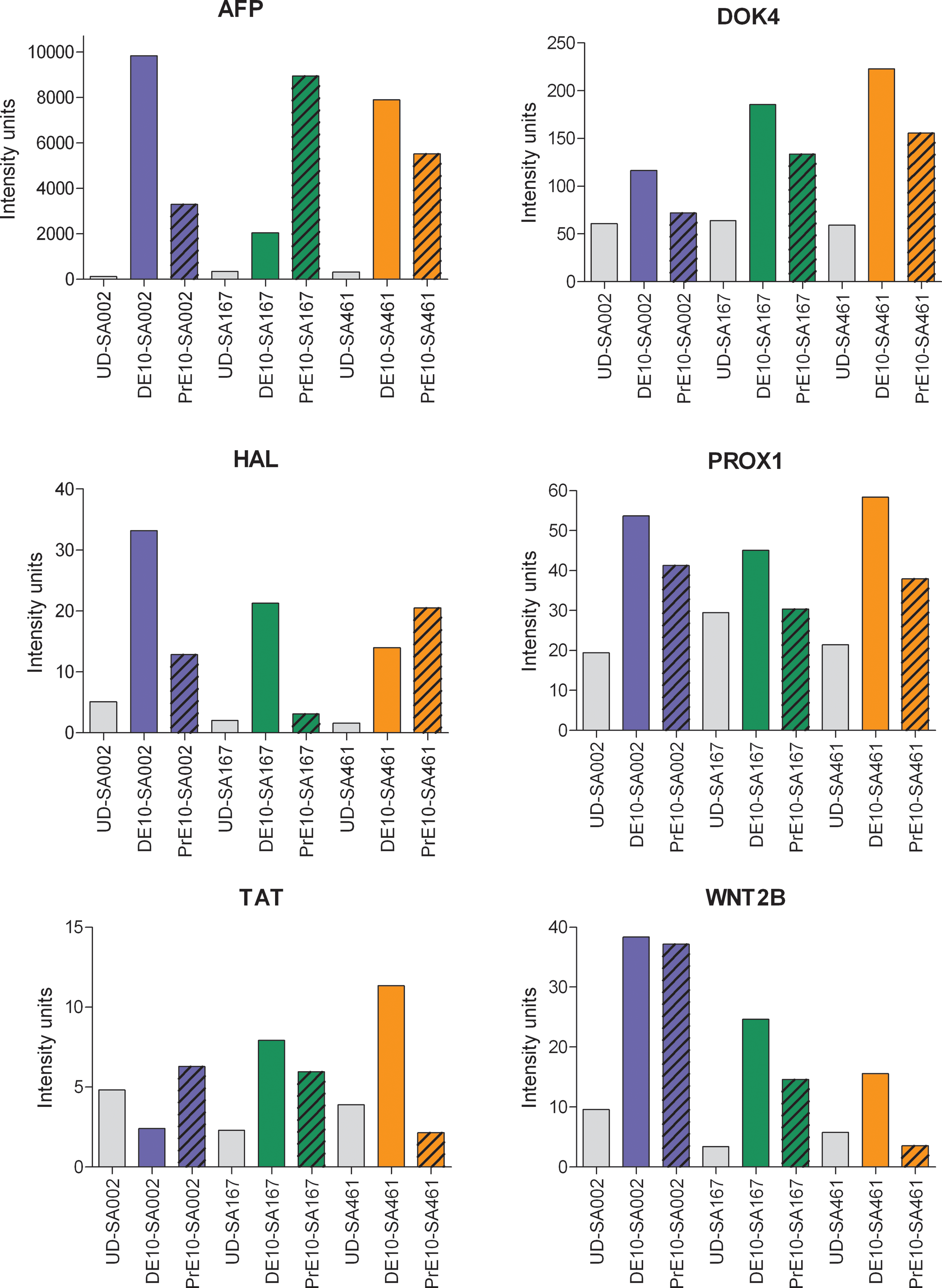
Marker genes for hepatocyte-progenitors. Gene expression levels for marker genes of early liver development in definitive endoderm (DE) and primitive endoderm (PrE) samples, harvested 10 days after onset of differentiation. Blue, green, and orange bars represent cell line SA002, SA167, and SA461, respectively, gray bars represent human embryonic stme cells (hESC). Solid bars represent samples where the DE-protocol was used and striped bars represent samples where the I-protocol was used. The intensity units are directly proportional to the mRNA level. Color images available online at
Moreover, marker genes expressed in adult hepatocytes such as the frequently reported ALB, DPP4, SERPINA7, TF [1,3,11,15,33,34] but also the less reported TM4SF1, SCL5A12, and UBD were monitored [35]. These genes demonstrate high expression in DE-Hep, while they typically display a blunted expression in the PrE derivatives, as shown in Figure 6. In addition the expression of CD44, which shows the highest levels in DE-Hep, is known to be expressed in hepatocyte-progenitors [36 –38].
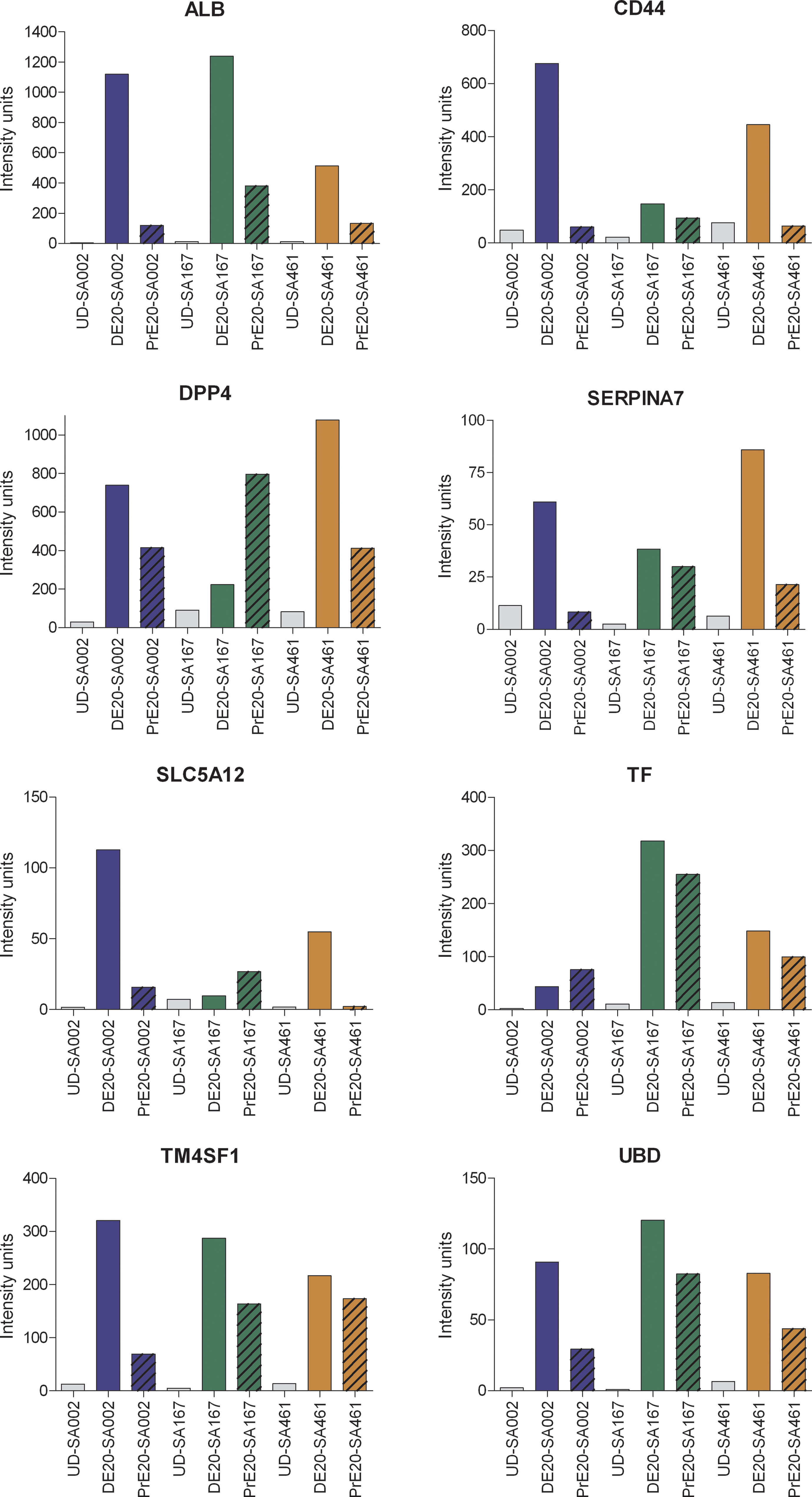
Marker genes for hepatocytes. Gene expression levels for genes known to be expressed in adult liver. Human embryonic stem cells (hESC) and samples harvested 20 days after onset of differentiation are shown in the figure. Blue, green, and orange bars represent cell line SA002, SA167, and SA461 respectively, gray bars represents UD. Solid bars represents samples where the DE-protocol was used definitive endoderm (DE) and striped bars represent samples where the I-protocol was used primitive endoderm (PrE). The intensity units are directly proportional to the mRNA level. Color images available online at
To get an indication of differences regarding functional properties of the cells differentiated using the DE-protocol versus the I-protocol, several cytochrome P450 (CYP) genes were monitored. Cells harvested at day 20 following the DE-and I-protocols were compared to UD samples and the HepG2 cell line (data not shown). Overall, only limited expression of CYP genes, similar to HepG2 cells, was detected in the hESC-derived hepatocyte-like cells irrespective of what protocol was used for the differentiation. However, in the DE-Hep samples up-regulated mRNA levels were observed across all the three investigated cell lines for some CYPs, known to act as important components in various metabolic functions, such as CYP1A1, CYP3A5, and CYP3A7 [3,14,33]. CYP1B1, a less studied CYP, was also up-regulated in DE-Hep across all three cell lines. In addition to the above CYPs, we also monitored the expression of CYP1A2, CYP3A4, and CYP7A1 in DE-Hep and in PrE derivatives, and compared their mRNA levels with those observed in UD cells and in HepG2. Each of these genes was up-regulated in DE-Hep compared to UD cells in one of the three cell lines: CYP1A2 and CYP3A4 were induced in SA002 and CYP7A1 was induced in SA461 (data not shown). Notably, CYP7A1, a marker claimed to be liver-specific [3,15,39], is not expressed in the corresponding PrE derivatives.
Comparison of up-regulated genes between the differentiation protocols
To identify genes that were exclusively up-regulated using the DE- and I-protocols, up-regulated genes with FC > 3 between UD samples and samples from each differentiation time point were summarized and compared. Notably, on the Affymetrix HGU 133 Plus 2.0 microarray each gene is represented by one or several probes, and the present subanalysis was conducted at the probe level. Therefore, some symbols may appear in more than one group due to, for example, variations in the gene expression between transcript variants. Figure 7 shows the number of up-regulated probes in each cell line and protocol, at each time point. In some cases, the number of unique genes represented by these probes in each group is given in parentheses. As shown in Figure 7A, 167 probes are up-regulated (FC > 3) in DE04 but not in PrE04, 42 probes are exclusively up-regulated in PrE04, and 4 probes are up-regulated in cells obtained from both differentiation protocols. The corresponding numbers of down-regulated probes are 50, 132, and 3, respectively. The number of up-regulated probes increases during the differentiation process. In the cells harvested on day 10 (Fig. 7B) 439 probes are exclusively up-regulated in the DE10 samples, 290 probes are up-regulated only in the PrE10 samples, and 282 probes are up-regulated in cells from both protocols. The corresponding numbers of down-regulated probes at day 10 are 243 for DE10, 67 for PrE10, and 59 for both protocols. Figure 7C represents day 20, which shows the largest number of differentially expressed probes when compared to UD cells. For this time point there were 921 up-regulated and 551 down-regulated probes in the DE20 samples only, while 257 probes were exclusively up-regulated and 168 were exclusively down-regulated in PrE20 samples. Moreover, 444 probes show up-regulation and 136 show down-regulation in the samples using any of the protocols. The complete lists of differentially expressed probes at each time point are available as Supplementary material (Supplementary table available, online at

Up-regulated genes in DE-derived hepatocyte-like cells (DE-Hep), and primitive endoderm (PrE) derivatives. Panel
Gene ontology annotation of genes up-regulated in DE-Hep
A gene enrichment analysis was conducted using Gene Ontology (GO) annotations in order to explore the up-regulated genes in DE-Hep according to their biological process (BP), molecular function (MF), and cellular component (CC) [23]. By applying a modified Fisher's exact test and using the whole human genome as a reference list, overrepresentation of annotations for the 941 up-regulated genes in DE-Hep was calculated. Significantly overrepresented annotations (P < 0.01) from all three categories: BP, MF, and CC were identified and the results show that annotations such as “regulation of cell proliferation,” “organ morphogenesis,” “tissue development,” and “cell differentiation” were significantly overrepresented in the BP category. In the MF category annotations such as “integrin binding” and “interleukin binding,” which are important in cell communication and cell differentiation, were significantly overrepresented. Moreover, in the CC category, “plasma membrane” and “collagen” are examples of the most significantly overrepresented annotations. Unexpectedly, “endoderm development” was not in the list of overrepresented annotations, but this can be explained by the fact that only two human genes are annotated with this GO-term. Therefore, all genes annotated as associated with “endoderm development” in any species were selected for further investigation. In total, 59 genes are annotated with “endoderm development,” and using homology search 37 corresponding human genes were found. The gene expression patterns of these 37 genes were explored in our data sets and the results demonstrate that 11 of these genes were up-regulated during DE development in all three cell lines. Interestingly, there is a striking difference in expression pattern between the differentiation protocols for these genes (Fig. 8).
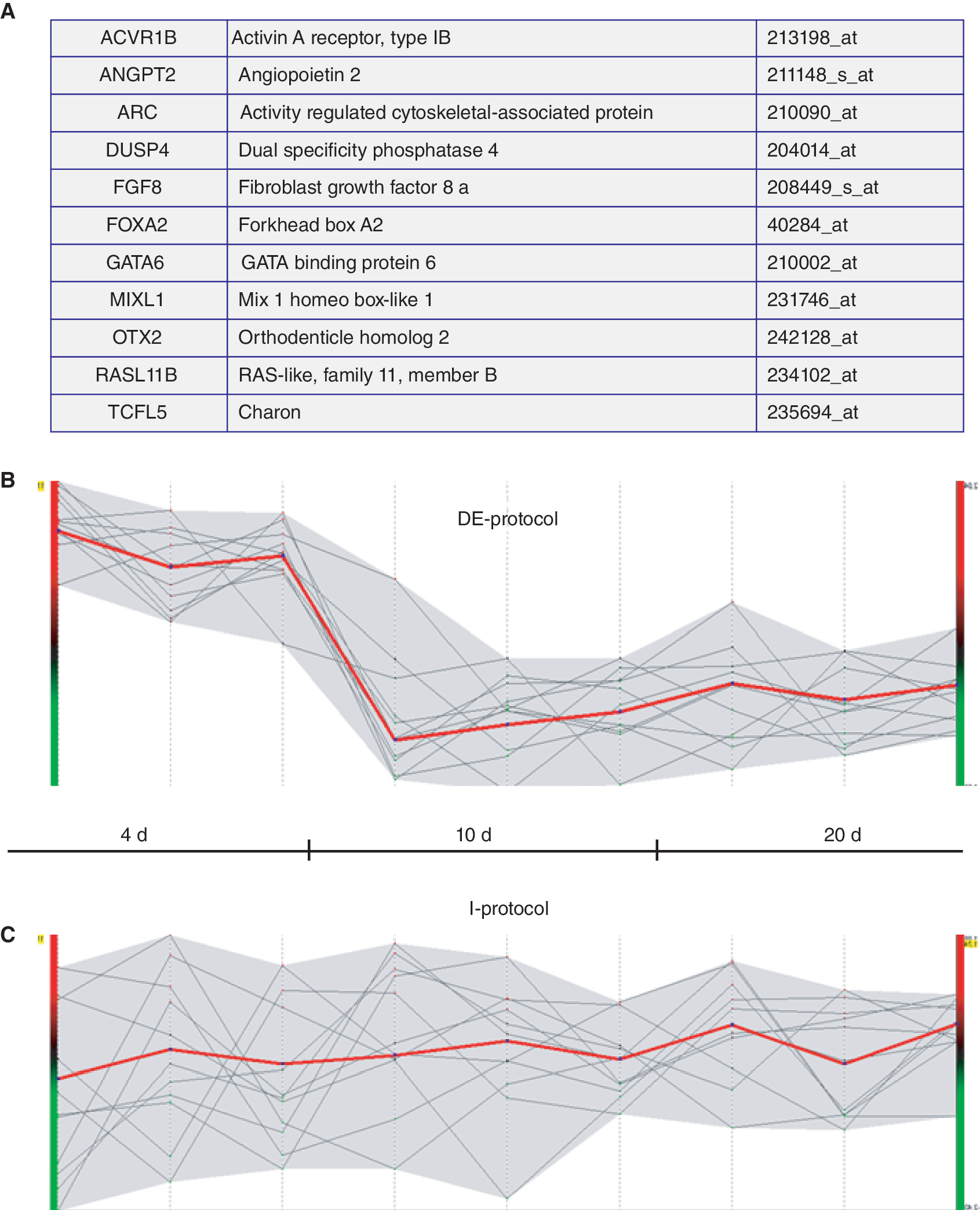
Expression profiles of “endoderm development” annotated genes. (
Up-regulated pathways in DE-Hep
The 941 up-regulated genes in DE-Hep were used as input to DAVID, and 850 of these genes were recognized by the tool. Furthermore, 284 genes were recorded in KEGG as involved in one or more cellular pathways. Using the whole human genome as a reference list and a modified Fisher's exact test, 11 pathways containing a significant (P < 0.05) number of the up-regulated genes were identified (Table 1).For comparison, an identical analysis was also conducted using the list of 515 up-regulated genes (FC > 3) in the PrE derivatives. Of these, 489 genes were identified by the DAVID tool, and 154 genes were recorded in the database as involved in one or more KEGG pathways. Using the same statistical test as above, 7 pathways containing a significant number of up-regulated genes in PrE derivatives were identified (Table 1), and 4 of them overlapped with the up-regulated pathways in DE-Hep.
KEGG pathways with a significant number of up-regulated genes in the DE-Hep and in the PrE derivatives. Overlapping up-regulated pathways across both differentiation regimes are italicized.
DE-Hep, DE-derived hepatocyte-like cells.
Protein interactions among up-regulated genes in DE-Hep and PrE derivatives
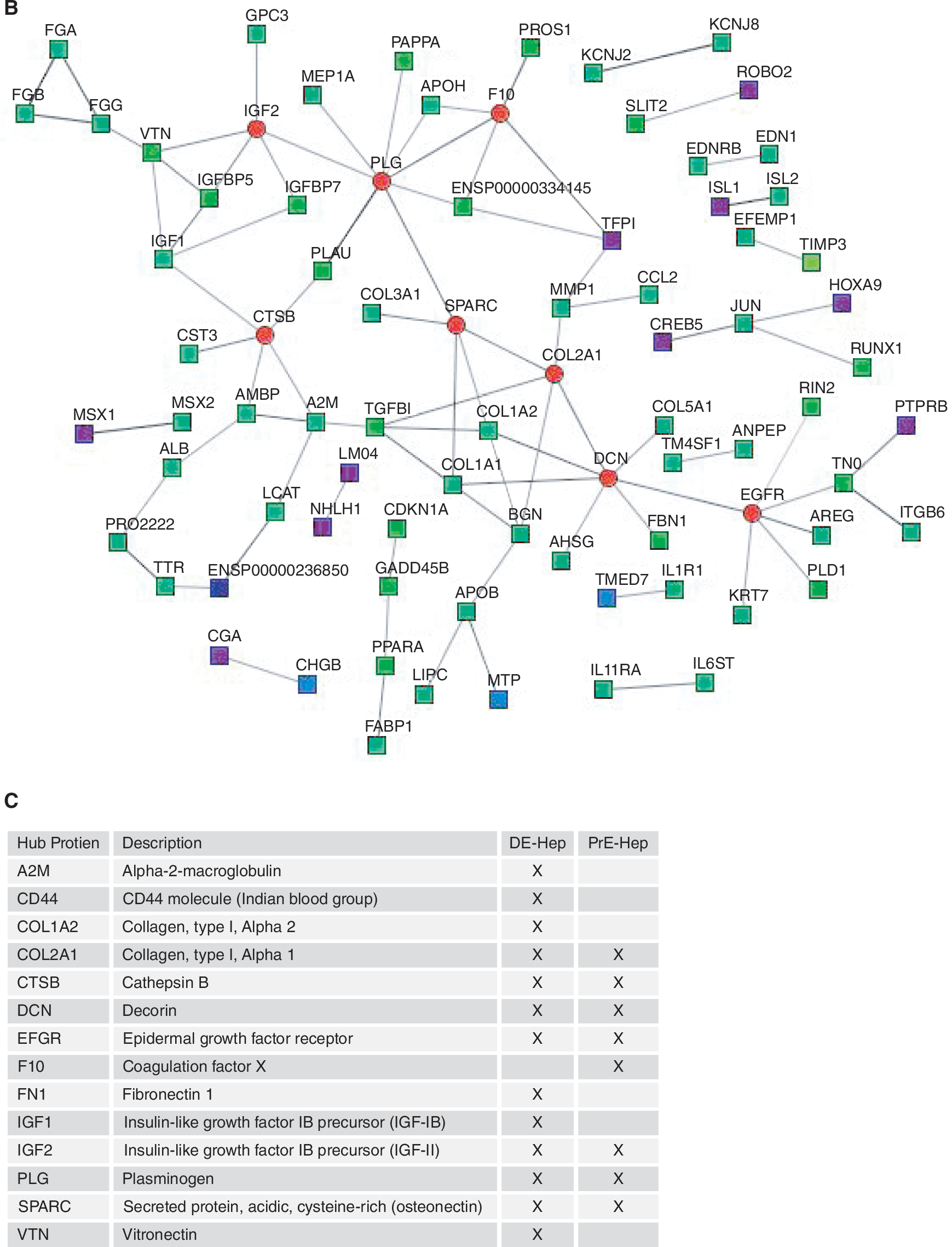
To explore protein interactions between the gene products from the up-regulated genes in the DE-Hep and the PrE derivatives, respectively, the STRING search tool was used to derive PINs. In order to achieve a high confidence in the PINs, only experimentally determined interactions were considered in this analysis. When analyzing the PINs, interesting topological characteristics were revealed and a number of hub proteins (proteins with ≥5 interactions) were identified (Fig. 9). In the set of 877 up-regulated genes in DE-Hep, 13 hubs were identified, whereas 8 were identified among the up-regulated genes in PrE derivatives. Six of the hubs (SPARC, IFG2, EFGR, DCN, PLG, and CTSB) overlapped across the protocols, whereas A2M, CD44, COL1A1, COL1A2, FN1, IGF1, and VTN are classified as hubs only in the DE-Hep, and F10 and COL2A1 are identified as hub proteins only in the up-regulated genes in PrE derivatives (Fig. 9).
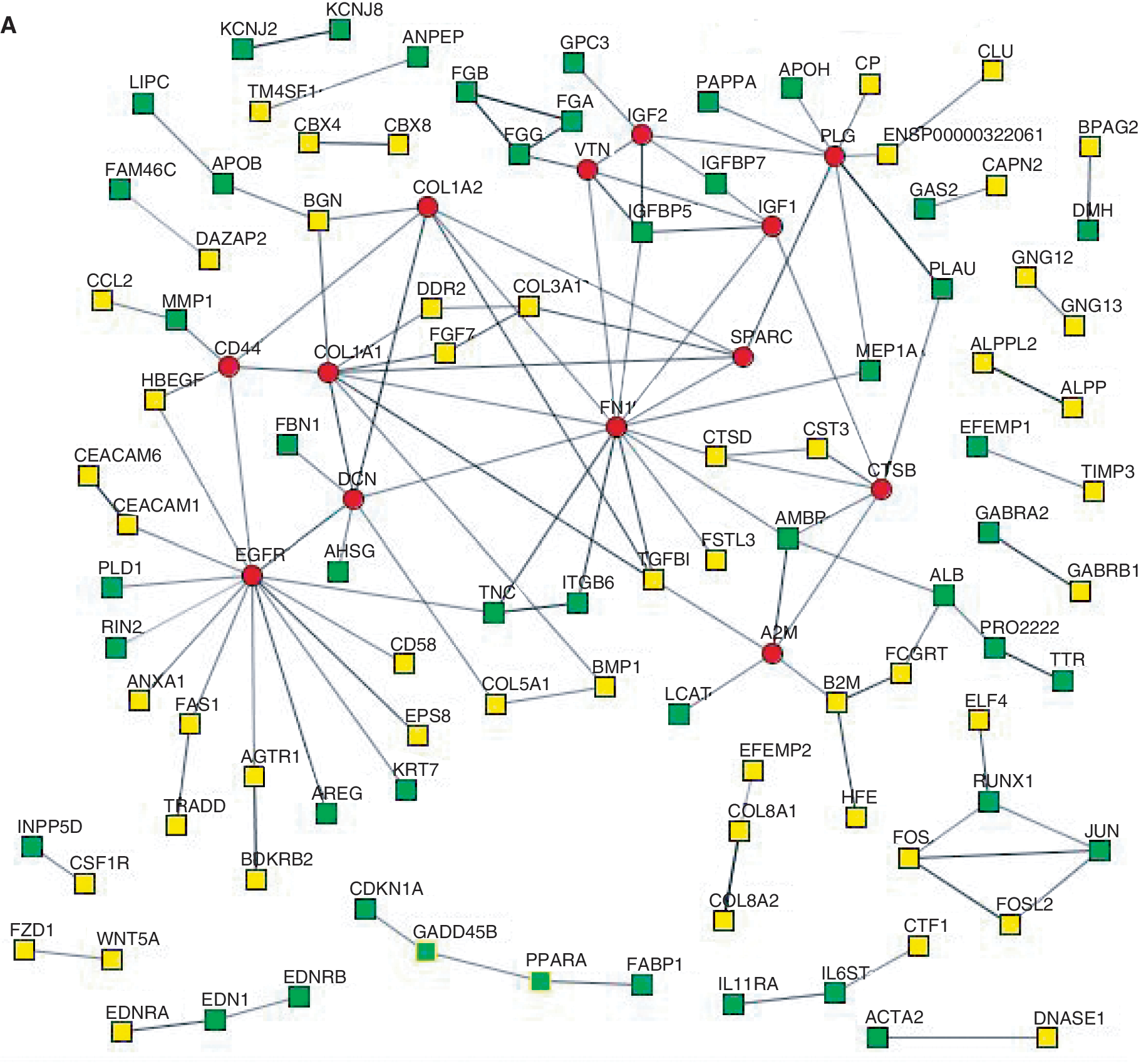
Protein interaction networks (PIN). Panels
Real-time quantitative PCR analysis
To corroborate the results from the microarray analysis, we selected a few of the markers from Figures 3 to 5 for further validation. The relative mRNA levels of POU5F1 (Oct4), CXCR4, SOX17, CER1, AFP, and ALB were determined in cell line SA002 at selected time points using real-time quantitative PCR. The results are shown in Figure 10 and confirm the expression profiles of these genes obtained in the microarray analysis.
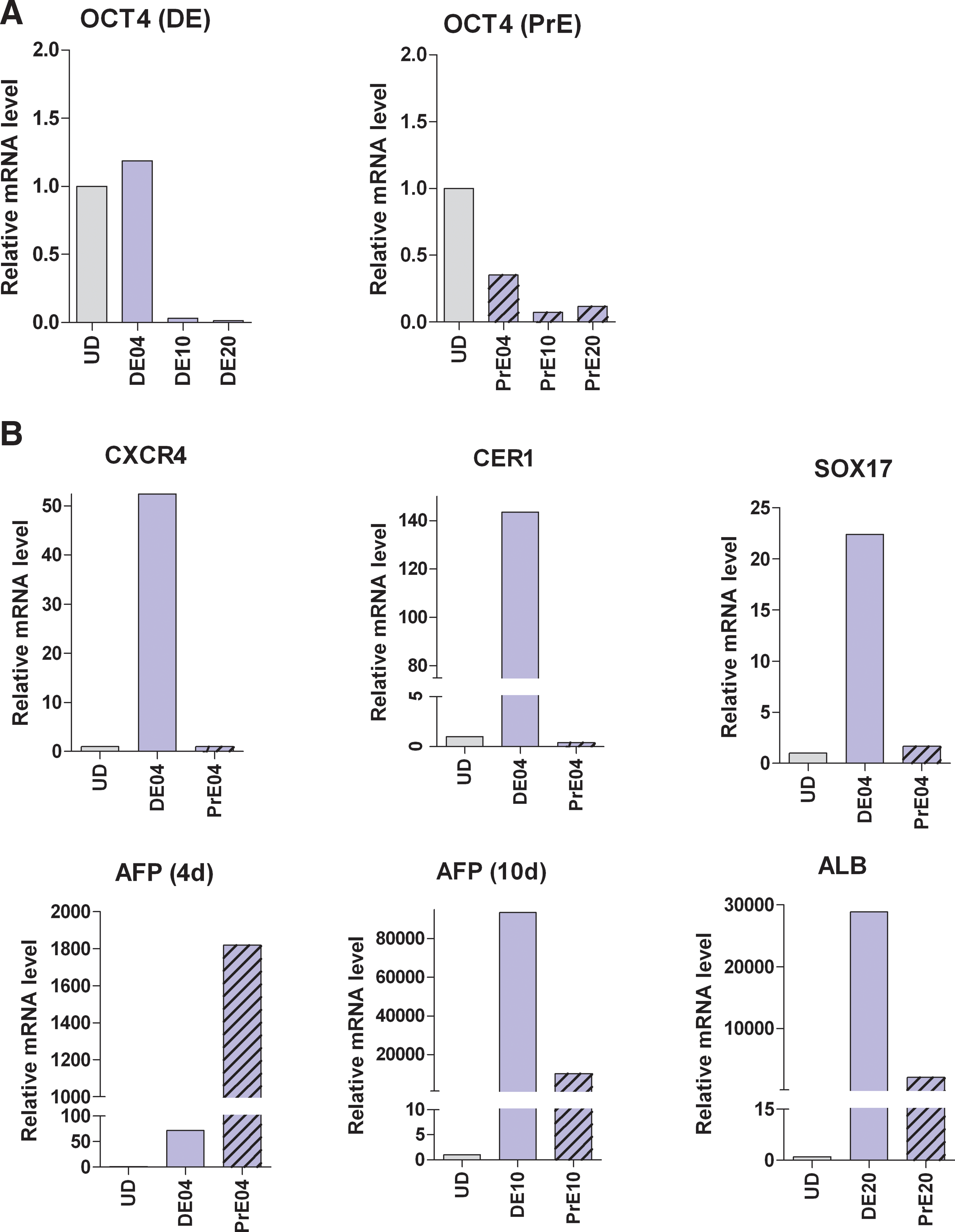
Real-time quantitative PCR. Panel
Discussion
Cell models based on hESC provide opportunities to study the molecular mechanisms regulating the development of the human hepatocyte lineage [41]. Differentiated hepatocyte-like cells represent promising tools for target evaluation, metabolism, and safety assessment of new drug candidates [42,43]. However, much more information is needed about the cues steering hESC to endoderm, hepaticprogenitors, and hepatocyte-like cells in order to optimize the properties of the resulting functional cells.
Microarrays provide powerful tools to study large-scale transcriptional activity. Using microarrays and various differentiation protocols, several investigators have reported on the capacity of hESC to differentiate into specific cell types. Until now these studies have mainly been conducted on derivatives from the mesoderm [28,44] and the ectoderm lineages [45], while to the best of our knowledge only one study [14] has reported on global transcriptional results from the endoderm lineage. In the present study we have, for the first time, investigated the global transcriptional profile of hESC derivatives primed through DE and PrE and validated the results by real-time qPCR and immunohisto-chemical analysis. We compared two differentiation protocols (the DE- and I-protocols) and cells were sampled at days 4, 10, and 20 after onset of differentiation (Fig. 1). The cells at these time points stain positive for key proteins known to be indicative for DE, hepatocyte-progenitors, and hepatocytes and the morphologies of the cells were characteristic [19]. An interesting transcriptional pattern was noted, and a much larger number of up-regulated genes was identified in the DE derivatives (yellow circles in Fig. 7) compared to the samples derived through the PrE (blue circles in Fig. 7). It was also clear from our results that the transcriptional activity increases dramatically as the cells differentiate. To investigate if the sets of genes that show up-regulation during the hepatic differentiation have been observed to be expressed in the liver in other studies, we explored their tissue expression patterns. These results show a very significant overlap (P values < 10−144) between up-regulated genes in our data and genes reported as overexpressed in liver tissue in other studies (Fig. 7D). The fraction of genes that show up-regulation in the DE derivatives and that are also overexpressed in liver tissue in public expression data increases as the cells differentiate. This indicates that our DE-protocol directs the hESC toward a cell population with a gene expression pattern that is highly overlapping with observations made from human liver tissue. In the present study our investigations focus on up-regulated genes, but down-regulation of genes may be equally important for stem cell differentiation. Further characterization of down-regulated genes is therefore of importance.
In Figure 3, the efficiency of the differentiation is demonstrated. The mRNA levels of four typical markers for pluripotency rapidly decrease as the cells differentiate. Twenty days after onset of differentiation, the expression levels for most of these genes were below, or close to, the detection level of the microarrays. We also investigated the expression levels of several known markers for DE, such as CXCR4, CER1, GSC, SOX17, FOXA2, and NODAL. All of these genes show very high expression in the DE04 samples compared to hESC, and to PrE derivatives at the corresponding time point. As demonstrated in Figure 4, CXCR4 and CER1 have several thousand units higher expression in DE04 than in PrE04. High expression of these markers in DE has also been reported by several other groups, both in human [3,8,30,31] and in mouse [6,13]. Notably, the hESC line SA002 showed substantially higher mRNA levels than SA167 and SA461 for all investigated DE04 marker genes. Both AFP and HNF4A, two genes known to be expressed in PrE [46 –48], display higher expression levels in samples from PrE than in samples from DE (Fig. 4B). These data are in line with the generation of DE and PrE as a result of the different protocols used.
The next differentiation stage to be investigated was the hepatocyte-progenitor stage, which is a challenging stage to define due to the lack of reliable markers for hepatocyte-progenitors [49]. A combination of markers is likely needed to discriminate hepatocyte-progenitors from other cell types. Here, we monitored a selection of six marker genes known to be important during hepatic initiation or expressed in immature hepatocytes [1, 50 –53]. Notably, DOK4, PROX1, and WNT2B show a consistent expression pattern across all investigated cell lines with a higher expression level in DE-derived hepatocyte-progenitors (DE-Prog) than in the corresponding samples derived from PrE (Fig. 5). Wnt signaling has been shown to be important in endoderm differentiation [2] and studies in zebra fish have shown that WNT2B positively regulates liver specification [54]. Also, AFP, HAL, and TAT are up-regulated in DE-Prog, and in two of the three cell lines studied here, these genes show the highest expression in the DE derivatives. AFP is well-studied and known to be expressed by immature hepatocytes [4,11,15], supporting the notion that this differentiation stage represents an early hepatic-progenitor stage.
Several markers for adult hepatocytes (ALB, DPP4, SERPINA7, SLC5A12, TF, TM4SF1, and UBD [4,11,14,33,35,36]) show increased mRNA levels in the DE-Hep samples (Fig. 6). Notably, also CD44, expressed in hepatocyte-progenitors [36 –38], show a high expression at 20 days, which indicates that the DE-Hep have an immature phenotype or also contain a fraction of hepatocyte-progenitors. Albumin is a well-established marker of mature hepatocytes [2,15,50,55], and 20 days after onset of differentiation ALB showed almost 1,000-fold higher expression levels in DE-Hep compared to UD samples. Notably, the expression of ALB was between 3 and 11 times higher in the DE-Hep than in the corresponding PrE derivatives (Fig. 6). Furthermore, SERPINA7, also a marker of hepatocytes [35], showed distinct up-regulation in DE-Hep. Of special note are the less reported TM4SF1 and UBD that demonstrate highly interesting expression patterns in our data. Their expression levels were consistent across all three cell lines, indicating a putative importance of controlled expression during hepatocyte differentiation. Both of these genes show increasing expression levels during the differentiation toward hepatocytes (data not shown), with the peak expression in the most mature samples (Fig. 6). Consistent with our observation, both of these genes have previously been reported as reliable hepatocyte markers [35]. Interestingly, the majority of the 8 genes in Figure 6 showed lower but significant expression levels in the corresponding PrE samples, indicating that the heterogeneous cell population derived using the I-protocol may contain a smaller fraction of cells with hepatic characteristics. The expression of several of these marker genes have also been verified with real-time QPCR and immunohistochemistry and the results are highly consistent with results from the microarray data [19].
CYPs are important for the metabolism of drugs and other xenobiotics in the liver. The expression levels of several CYP genes were therefore monitored in the DE-Hep. CYP7A1 is liver-specific and it is not expressed in the yolk sac [3,15,39]. Notably, CYP7A1 was induced in DE-Hep samples from SA461 but not in the corresponding PrE derivatives, supporting the hypothesis that the DE-Hep share characteristics with the adult human liver. Moreover, we observed high expression of CYP1B1 in the DE-Hep samples compared to hESC in all three cell lines. This gene was also measured during hESC differentiation to hepatocytes by Duan et al. [33], and their results show expression of CYP1B1 both in vitro and in vivo. In our data, the expression of this gene increased gradually during the hepatic differentiation (data not shown). Notably, the expression levels of all CYPs are in the same range, or higher, compared to HepG2 cells (data not shown).
Besides analyzing and interpreting the results from the expression profiling for genes individually, we also defined protein interaction maps for the up-regulated genes in hESC-derived DE-Hep and the corresponding PrE derivative (Fig. 9). This protein interaction analysis revealed seven hub proteins that were exclusively identified in DE-Hep (A2M, CD44, COL1A1, COL1A2, FN1, IGF1, and VTN). Interestingly, these proteins have, in other organisms, been shown to play important roles in liver and hepatocyte development. The ortholog of A2M in zebra fish was recently reported to be essential for liver cell differentiation and tissue development [56]. The A2M family proteins are also known to be mainly produced in the liver and Hong et al. [56] showed that A2M is expressed in endodermal precursors and subsequently in the liver. By knockdown studies, they demonstrated that A2M is required for liver formation, specifically for cell proliferation in the liver rudiment. The CD44 gene encodes a family of alternatively spliced adhesion molecules and it has been suggested to be a marker of rat hepatic-progenitor cells [36]. CD44 appears to have dual roles and may be involved in proliferation and maintenance of hepatocyte-progenitors, but it is also associated with maturation of hepatocytes. As suggested by Roskams [49], important future work will be to study the functional aspects of CD44 expression in human liver. Our results demonstrating that CD44 is a hub protein in DE-Hep are an important piece of information indicating that CD44 may be important for hepatocyte differentiation also in humans. Among the potentially interesting hub proteins in DE-Hep are also the insulin-like growth factors including IGF1 and IGF2, which are of major importance for normal growth and development [57]. The mRNA levels of IGF1 in the liver are 10- to 100-fold higher than in the majority of other tissues [57]. Moreover, several studies of vitronectin support our results that suggest an important role of VTN expression in DE-Hep samples. VTN is expressed in several tissues, but predominantly in the liver [58].
Using the KEGG pathway database, we identified several pathways that show significant up-regulation in DE-Hep and PrE derivatives compared to hESC. Some of these pathways, such as “cell communication,” “ECM–receptor interaction,” and “focal adhesion” are known to be important for adhesive interactions between cells and their microenvironment, which is critical for cell-fate determination and differentiation [59]. It was therefore not unexpected to find these pathways up-regulated. Moreover, the “gamma-hexachlorocyclohexane degradation” pathway is exclusively up-regulated in DE-Hep. This pathway is classified in KEGG as involved in metabolism of xenobiotics and thus closely related to functions of the liver. Central in all aspects of cell-fate determination are the integrins, and our results demonstrate up-regulation of many integrins (Supplementary table).
Our study demonstrates substantial transcriptional differences between DE and PrE differentiation even if the effects of these differences at protein levels require further investigation. Clear expression patterns resembling hepatic development were observed only in the DE derivatives. In these samples we also identified less variation between cell lines than in the PrE, similar to previous observations that cell line variability is affected by the differentiation protocols [60]. However, as the cells differentiate further, some variations between cell lines become apparent. With a few exceptions, most of the hepatic markers show distinct up-regulation in the DE-Hep, indicating that this population comprises hepatocyte-like cells that may be immature but nevertheless committed toward the hepatic lineage. Therefore, in future studies it would be interesting to also include differentiation time points later than 20 days and further investigate the maturation potential of committed cells. The observed variation between the cell lines may be due to the fact that differentiated cultures are heterogeneous, and in particular the PrE derivatives contain a mixture of cells that probably varies to some extent between the samples. Another possibility is that the rate of differentiation may vary between the cell lines, that is the cell populations are not synchronized at every differentiation stage. In agreement with these arguments are other studies reporting on large variation between hESC lines [16,17]. Thus, our results provide a foundation for subsequent studies of specific factors and pathways that could induce hepatic differentiation from hESC in multiple cell lines.
In conclusion, we performed global gene expression analysis of hESC-derived DE and PrE, which were also further differentiated toward the hepatic lineage. Differentiated cells were harvested at discrete points of time after onset of differentiation, and we identified large sets of exclusively upregulated genes in each of the sampled cell populations. We monitored markers for DE, PrE, hepatocyte-progenitors, and hepatocytes, and the latter two populations of cells showed typical expression patterns for hepatic differentiation, especially in the DE derivatives. Interestingly, seven hub proteins and 7 significantly up-regulated cellular pathways were exclusively identified in the DE-Hep. Several of these hub proteins and pathways are associated with liver or with hepatic differentiation. Taken together, our results provide a comprehensive characterization of hESC-derived endoderm, hepaticprogenitors, and hepatocytes at the transcriptional level and reveal novel key genes and pathways of potential importance for future efforts to improve hepatic differentiation.
Footnotes
Acknowledgments
The studies presented herein were financially supported by the Information Fusion Research Program (University of Skövde, Sweden) under grant 2003/0104 from the Knowledge Foundation, and the EU-funded projects Vitrocellomics (contract no. LSHB-CT-2006-018940) and Carcinogenomics (contract no. LSHB-CT-2006-037712). We acknowledge the production team at Cellartis for the supply of human embryonic stem cells and the Microarray Resource Centre at Lund University for their help with the microarray experiments.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.