
Abstract
Multipotent mesenchymal stromal cells (MSC) are present in bone marrow and other tissues such as adipose tissue, muscle, pancreas, liver, and so on. Recent evidence suggests that MSC migrate to sites of infection, inflammation, and cancer, and interact with different immune cell subsets. Here, we report for the first time on the isolation and characterization of multipotent nasal mucosa-derived mesenchymal stromal cells (nm-MSC). nm-MSC showed a plastic adherent and fibroblast-like morphology and were able to form colonies. They expressed the typical bone marrow MSC marker antigens CD29, CD44, CD73, CD90, and CD105 and were able to differentiate along the adipogenic, chondrogenic, and osteogenic pathways. nm-MSC produced a set of inflammatory cytokines, expressed chemokine receptors, and were responsive to stimulation with cytokines, chemokines, and TLR4 ligand LPS. Thus, these cells may serve as an alternative adult stromal cell resource for regenerative tissue repair and may represent important regulators of local mucosal immunity.
Introduction
T
There is evidence that MSC do not only exist in bone marrow, but also in many other tissues and organs, including muscle, bone, cartilage, tendon, adipose tissues, umbilical cord blood, heart, salivary gland, liver, and the synovial membrane [4 –8]. The identification of such multipotent cell populations from different tissues has increasingly attracted attention in the last few years.
In addition to being an attractive tool in cell-based medicine, recent evidence suggests a role for MSC in modulating local immunity [9]. MSC express cytokine and chemokine receptors along with adhesion molecules frequently found on classical immune cells. MSC interact with various immune cell subsets and suppress immune responses in vivo [10 –12]. Thus, it appears that MSC may play a central role in local immune regulation. The isolation and characterization of tissue-resident MSC is important not only for development of novel cell-based therapeutic approaches, but also for understanding local tissue immune regulation. MSC of the mucosa of the upper respiratory tract have not been defined thus far. The present manuscript is the first to report the isolation and characterization of MSC from nasal mucosa (nm-MSC). We show that nm-MSC express a set of surface antigen markers also found on bone marrow-derived mesenchymal stromal cells (bm-MSC), are capable of long-term expansion in vitro, and show typical trilineage differentiation [3].
Initial immunological studies demonstrate that these cells produce a set of inflammatory cytokines, express chemokine receptors, and are responsive to immunological signals. These cells may be an alternative adult stromal cell resource for regenerative tissue repair and autotransplantation in clinical approaches and may represent important regulators of local mucosal immunity.
Materials and Methods
Tissue specimens, cell preparation, and culture conditions
Nasal mucosa specimens of the inferior nasal concha (usually <1 g) were obtained from patients undergoing reduction of the nasal turbinate at the Department of Otorhinolaryngology, University Hospital Essen, Germany. The patients (n = 13) were 23–48 years old and gave an informed consent. The study was approved by the local ethics committee (No. 08-3710).
After resection, tissue samples were collected aseptically in NaCl (0.9%; Fresenius Kabi, Bad Homburgh, Germany). Subsequently, nasal mucosa specimens were washed several times with Ringer's solution (Braun, Melsungen, Germany) in order to remove the majority of erythrocytes. Tissues were cut into 1–2 mm pieces, washed extensively, and digested in Ringer's solution containing 5.01 mg/mL collagenase type II (Worthington Biochemical Corporation, Lakewood, NJ) and 3.99 mg/mL hyaluronidase (Worthington Biochemical Corporation, lakewood, NJ, USA) for 40 min at 37°C with gentle shaking. Tissues were centrifuged at 1,200 rpm for 7 min, and the supernatant was discarded. The partially digested tissues were further treated with Ringer's solution and 33.4 mg/mL dispase (Roche Applied Science, Mannheim, Germany) for 60 min at 37°C.
The cell suspension was then centrifuged and the pellet was resuspended in standard culture medium (high-glucose DMEM (PAA, Pasching, Austria) supplement with 10% fetal bovine serum (FBS; Biochrom, Berlin, Germany), 1% penicillin/streptomycin (PAA, Pasching, Austria), 1% sodium pyruvate (PAA, Pasching, Austria) and transferred to tissue culture flasks. Non-adherent cells were removed by washing with phosphate-buffered saline (PBS) 48 h later and fresh medium was added over the remaining cells. During the culture period, cells were maintained at 37°C in a humidified atmosphere of 5% CO2. Cells were continuously passaged after reaching subconfluency by Accutase (PAA, Pasching, Austria) treatment for 5 min at 37°C.
Colony-forming unit fibroblast (CFU-F) assay
The CFU-F assay was performed by plating 0.5 × 106 cells of the initial tissue single cell suspension in standard culture dishes (Sarstedt, Nümbrecht, Germany) in standard culture medium. Medium was exchanged after 48 h and later on every 3–4 days. After 14 days in culture, cells were washed twice with PBS and fixed with 10% formalin for 20 min at room temperature. To visualize and count MSC-CFU, cells were stained with Giemsa (Sigma-Aldrich, Taufkirchen, Germany) and air-dried for 5 min. CFU colonies between 5 mm and 8 mm in diameter were scored macroscopically.
Cell doubling time
Cells between passages 1 and 10 were plated in standard culture medium at a density of 5 × 106 cells/cm2 in T25 culture flasks. Standard culture medium was changed every 3 days until the adherent cell population reached 80% confluence. Cells were continuously passaged after reaching subconfluency by Accutase (PAA, Pasching, Austria) treatment for 5 min at 37°C. Cell doubling time (DT) was calculated for each passage according to the following formula:
CD = ln(N f/N i)/ln(2)
DT = CT/CD
where DT is cell doubling time; CT, cell culture time; CD, cell doubling number; N f, final number of cells; and N i, initial number of cells [13].
Flow cytometry
To evaluate cell-surface marker expression, cells were collected and 0.2–0.5 × 106 cells were placed into FACS tubes (BD Bioscience, Heidelberg, Germany). Cells were incubated for 30 min at 4°C with 5 μL conjugated or 5 μL unconjugated monoclonal antibodies specific for human markers associated with mesenchymal and hematopoietic lineages. To determine nonspecific signals, isotype controls were used at the same concentration used for the specific antibody. The following antibodies were used: CD11b (integrin αM, CR3, clone: clone Mac-1; BD Bioscience, Heidelberg, Germany), CD14 (LPS receptor, clone: M5E2; BD Bioscience), CD29 (β1-integrin, clone: MAR4; BD Bioscience), CD31 (PECAM-1, clone: WM59; BD Bioscience), CD34 (My10, clone: 581 (Class 3), Invitrogen, Karlsruhe, Germany), CD44 (H-CAM, clone: 515; BD Bioscience), CD45 (leukocyte common antigen (LCA), clone: 5B1; Miltenyi Biotec, Bergisch Gladbach, Germany), CD54 (ICAM-1, clone: 15.2; Chemicon, Limburg, Germany), CD71 (transferrin receptor, clone: AD2; BD Bioscience), CD73 (ecto-5-NT, SH4; clone: AD2; BD Bioscience), CD90 (Thy-1, clone: 5E10; BD Bioscience), CD105 (endoglin/TGF (1-b3 receptor, clone: 166707; R&D, Minneapolis, MN), CD106 (V-CAM-1, clone: 51-10C9; BD Bioscience), CD117 (c-kit, clone: YB5.B8; BD Bioscience), CD271 (low-affinity nerve growth factor receptor, clone: C40-1457; BD Bioscience), and STRO-1 (clone: STRO-1; R&D). Analysis was performed using a FACSCalibur Flow Cytometer (BD Bioscience) and the resulting data were processed using WinMDI 2.9 software (Windows Multiple Document Interface for Flow Cytometry).
Osteogenic differentiation
For osteogenic differentiation, we used osteogenic induction medium (standard culture medium supplemented with 0.06 mg/mL ascorbic acid (Sigma-Aldrich, Taufkirchen, Germany), 0.1 μM dexamethasone (Sigma-Aldrich), and 10 mM 3-glycerophosphate (Sigma-Aldrich, Munich, Germany).
Cells were seeded at a density of 3 × 103 cells/cm2 on round glass slides in 12-well culture dishes (Greiner Bio-One, Frickenhausen, Germany), until reaching 75% confluency, and were fed every 3–4 days with osteogenic induction medium for 2 weeks. Osteogenic differentiation was detected either by immunofluorescence after fixation with 10% formalin (Merck, Darmstadt, Germany) for 15 min at room temperature or by RT-PCR analysis.
Adipogenic differentiation
Differentiation was performed as described previously [13]. In brief, cells were seeded at a density of 3 × 103 cells/cm2 on round glass slides in 12-well culture dishes, cultured in standard culture medium until reaching 95% confluence. Adipogenic differentiation was induced by culturing cells in standard culture medium containing 2 μM insulin (Sigma-Aldrich, Taufkirchen, Germany), 500 μM 3-isobutyl-1-methyl-xanthine (IBMX; Sigma-Aldrich), 1 μM dexamethasone (Sigma-Aldrich), and 200 μM indomethacin (Sigma-Aldrich) for 3 days. This medium will be referred from here on as “induction medium.” Subsequently, cells were cultured in standard medium containing 2 μM insulin (maintenance medium) for 3–4 days. The cycle was repeated 3 times.
In order to examine the generation of oil droplets in the cytoplasm after differentiation, cells were fixed with 10% formalin (Sigma-Aldrich) and stained with Sudan III (Sigma-Aldrich) for 60 min at room temperature. Hematoxylin (Roth, Karlsruhe, Germany) was used to visualize nuclei. Additionally, the adipogenic differentiation was verified by RT-PCR analysis.
Chondrogenic differentiation
To induce chondrogenic differentiation, 2.5 × 106 cells were transferred into a 15-mL polypropylene tube and centrifuged at 1,000 rpm for 5 min to form micromass pellets at the bottom of the tube. After 48 h of culture in standard medium, chondrogenic differentiation was induced with standard medium supplemented with dexamethasone: 1 × 10−;3 M
Cytochemistry, histochemistry, immunohisto-chemistry, and immunocytochemistry
The adipogenic and osteogenic differentiated cells were grown on round glass slides in 12-well culture dishes for 21 days, washed in PBS, and fixed in ice-cold methanol–acetone (1:1). Micromass bodies induced for chondrogenic differentiation were embedded after 4 weeks of cultivating in tissue tec (Sakura Finetek, Staufen, Germany), cryosectioned at 5 μm thickness, and frozen at −;20°C. For detection of osteogenic or chondrogenic differentiation, we used monoclonal antibodies against osteopontin (Santa Cruz Biotechnology, Santa Cruz, CA, diluted 1:20) and collagen type II (Abcam, Cambridge, UK, diluted 1:50). After 3 washing steps with PBS, unspecific antibody binding was blocked by preincubation with 5% BSA (PAA, Paching, Austria) followed by incubation with monoclonal antibodies for 1 h. For immunofluorescence staining 7-aminoactinomycin (7-AAD; BD, Heidelberg, Germany) was used to counterstain cell nuclei. Samples incubated with 1% BSA only served as negative control. To demonstrate the presence of glycosaminoglycans in specimens induced for chondrogenic differentiation, we used Alcian blue staining. Dried cryosections were fixed with formalin and washed with PBS. Staining with Alcian blue 8GX (Roth, Karlsruhe, Germany) was performed at room temperature for 60 min and Nuclear Fast Red (Merck, Darmstadt, Germany) was used for counterstaining nuclei.
As a marker for osteogenic differentiation, alkaline phosphatase staining was performed. In brief, cells were washed with PBS, fixed in acetone–formalin in citrate, and washed with distilled water. Cells were incubated with sodium nitrite and FRV–alkaline phosphatase solution (Sigma-Aldrich, Taufkirchen, Germany) with naphthol-AS-BI alkaline solution (Sigma-Aldrich) for 15 min at room temperature in the dark. Hematoxylin (Merck, Darmstadt, Germany) was used for counterstaining nuclei.
Alizarin red S staining was performed to detect microcrystalline or noncrystalline calcium phosphate salts. After fixation with formaldehyde, cells were stained with 1% Alizarin red S (Sigma-Aldrich). After 5 min, red–orange staining of calcium could be observed microscopically.
The adipogenic differentiation was observed by staining with Sudan III to show the presence of fatty vacuoles in the monolayer culture. In brief, cells were washed twice in PBS, and 0.03% Sudan III in 10 mL 70% ethanol was added. Hematoxylin was used to counterstain nuclei in immunohistochemical stainings. Cells were analyzed with a standard or fluorescence microscope (Axioskope 2; Zeiss, Jena, Germany).
Conventional and quantitative real-time polymerase chain reaction
For conventional and quantitative polymerase chain reaction (PCR) analysis, total RNA was isolated from cultured cells as well as from the micromass bodies using the RNeasy Mini kit (Qiagen, Hilden, Germany). For detection of chemokine receptors, cells were preincubated for 16 h with or without interferon-gamma (IFN-γ, 400 U/mL; Peprotech, Hamburg, Germany) or lipopolysaccharide (LPS, 100 ng/mL, Microbiology and Infection Biology, Biophysics, Research Center Borstel) or macrophage migration inhibitory factor (MIF, 20 ng/mL; R&D Systems, Minneapolis, MN). RNA concentrations were determined by measuring the absorbance at 260 nm. Samples of RNA were reversely using oligo-dT primer and Superscript II reverse transcriptase according to the manufacturer's instructions (Invitrogen, Karlsruhe, Germany). Synthesized cDNA was used as template for the RT-PCR analysis. For real-time PCR, cDNA was pooled from 3 patients. RT-PCR was conducted using primers specific for osteogenic, adipogenic, and chondrogenic differentiation genes. The following sets of primers were used: aP2 (fatty acid-binding protein), sense 5′-GCTTTGCCACCAGGAA AGTG-3′ and antisense 5′-ATGACGCATTCCACCAC CAG-3′; C/EBP (CCAAT/enhancer-binding protein α), sense 5′-AGA AAGGGGTGGAAACATAGG-3′ and antisense 5′-GAAAGCTGAGGGCAAAGG-3′; osteopontin, sense 5′-ACTGATTTTCCCACGGACCT-3′ and antisense 5′-CATTCAACTCCTCGCTTTCC-3′; osteocalcin, sense 5′-CTCACACTCCTCGCCCTATT-3′ and antisense 5′-CGCCTGGGTCTCTTCACTAC-3′; collagen II, sense 5′-AGGCTCCCAGAACATCACCT-3′ and antisense 5′-ACAGTCTTGCCCCACTTACC-3′; aggrecan, sense 5′-TCAGGAACTGAACTCAGTGG-3′ and antisense 5′-GCCACTGAG TTCCACAGA-3′. Human β-2M (2-microglobulin), sense 5′-AGCGTACTCCAAAGATTCAGGTT-3′ and antisense 5′-ATGATGCTGCTTACATGTCTCGAT-3′ was utilized as positive control.
PCR cycles were as follows: initial denaturation at 95°C for 2 min, followed by 35 cycles of 95°C for 40 s, annealing at the primer-specific temperature for 40 s, 72°C for 40 s, and final extension at 72°C for 5 min.
Quantitative real-time PCR (Light cycler, Roche) was conducted using primer-specific chemokine receptor genes. Following sets of primers were used: CXCR1, sense 5′-CGCCAGGCTTACCATCCA-3′ and antisense 5′-CAAACAGCGGCACGATGA-3′; CXCR2, sense 5′-GAGGTGTCCTACAGGTGAAAAGC-3′ and antisense 5′-GGGGGCAGGGTAGAGCTGTA-3′; CXCR3, sense 5′-TGGCCGAGAAAGCAGGG-3′ and antisense 5′-AGGCGCAAGAGCAGCATC-3′; CXCR4, sense 5′-GGCCGACCTCCTCTTTGTC-3′ and antisense 5′-GGACGATGGCCAGGTAGC-3′; CXCR5, sense 5′-TTGCCTTGCCAGAGATTCTCTT-3′ and antisense 5′-TGAACCAGGCATGCGTTTC-3′; CXCR6, sense 5′-CATCTCTGCTGGTGTTCATCAGA-3′ and antisense 5′-GAACTGCAGGAAGTCTTGATGCT-3′; CXCR7, sense 5′-TCTTCTCCTACGTGGTGGTCTTCC-3′ and antisense 5′-GACAGGCACTGTGTGACATGC-3′; CCR1, sense 5′-ATAGAGAGGGAATGTAATGGTGGCC-3′ and antisense 5′-CCAAGCCCTTCTCTGGTACACTTAGT-3′; CCR2, sense 5′-CTGAGACAAGCCACAGCTGAAC-3′ and antisense 5′-CCGCTCTCGTTGGTATTTCTGA-3′; CCR3, sense 5′-CCGGTGATCTACGCCTTTGTT-3′ and antisense 5′-CAGCTTCTCACTAGGAAGGAATGG-3′; CCR4, sense 5′-AAAGCAAGCTGCTTCTGGTTG-3′ and antisense 5′-TTGGTGCAAGGCTTGGG-3′; CCR5, sense 5′-ACAAGAAACTCTCCCCGGGT-3′ and antisense 5′-GAACACCAGTGAGTAGAGCGGA-3′; CCR6, sense 5′-CCCACAATGAGCGGGGA-3′ and antisense 5′-CAAATAGCCTGGAGAACTGCCTG-3′; CCR7, sense 5′-ATTTTCCAGGTATGCCTGTGTCA-3′ and antisense 5′-TGATGGAGTACATGATAGGGAGG-3′; CCR8, sense 5′-CACTAAGGTCCCGCTGCCT-3′ and antisense 5′-GGCAATAAAAGACAGCAAGGAG-3′; CCR9, sense 5′-GCATCTGACTGACCCACCAT-3′ and antisense 5′-GCCTGACATTGTTTTTCTCACAGTAG-3′; CCR10, sense 5′-GAGGCCACAGAGCAGGTTTC-3′ and antisense 5′-CCTGGACATCGGCCTTGTA-3′; CX3CR1, sense 5′-CCAGGCCTTCACCATGG-3′ and antisense 5′-CAGGCCAATGGCAAAGATG-3′; XCR1, sense 5′-ACCATGGCTGTGTGGGTAGCC-3′ and antisense 5′-TCCCCAGGGACAGCAGGA-3′.
The conditions for amplification were: 5 min at 95°C, followed by 50 cycles of 30 s at 95°C, 30 s at 58°–64°C (according to primer), 40 s at 72°C, followed by an extension for 4 min. Amplified PCR products were separated by 2% agarose gel electrophoresis and the bands were visualized by ethidium bromide.
Enzyme-Linked Immunosorbent Assay (ELISA) for detection of cytokines
Secretion of IL-6, IL-8, IL-10, TNF-α, IFN-γ, granulocyte colony-stimulating factor (G-CSF), or MIF was detected by DuoSet Human Immunoassay (R&D Systems, Wiesbaden, Germany), according to the manufacturer's protocol. Third-passage stromal cells were washed 3 times with DMEM and cultured for 48 h in standard medium at a concentration of 1 × 106 cells/mL. Cells were incubated with or without the following factors: SDF-1α (stromal cell-derived factor-1α; concentration 100 μg/mL), TNF-α (1,000 U/mL), IFN-γ (400 U/mL), and the combination of TNF-α (1,000 U/mL), and IFN-γ (400 U/mL; all from Peprotech, Hamburg, Germany) for 16 h. Supernatant samples were run in triplicate and compared to standard curves of IL-6, IL-8, IL-10, TNF-α, IFN-γ, and MIF.
Results
Isolation of adherent colony-forming cells with fibroblastic morphology from nasal mucosa
Firm adherence to plastic is a key feature of MSCs. To test whether nasal mucosa tissue can be a source of such multipotent cells, nasal mucosa-derived fibroblast-like cells from 13 adult patients were isolated, plated, and cultured in standard culture medium. After 2 days, cultures of digested nasal mucosa tissue in the standard culture medium showed that the majority of the cells or cell aggregates remained in suspension. After extensive washing, few attached plastic adherent single cells were observed, which formed an adherent monolayer after 2–3 weeks of culture. These adherent cells proliferated actively and became nearly confluent. Morphologically, these putative nasal mucosa-derived MSCs were of serpiginous or fibroblast-like (Fig. 1) morphology similar to previously reported adipose- or bone marrow-derived mesenchymal stromal cells. This morphology was maintained through the whole culture period (to date >10 passages).
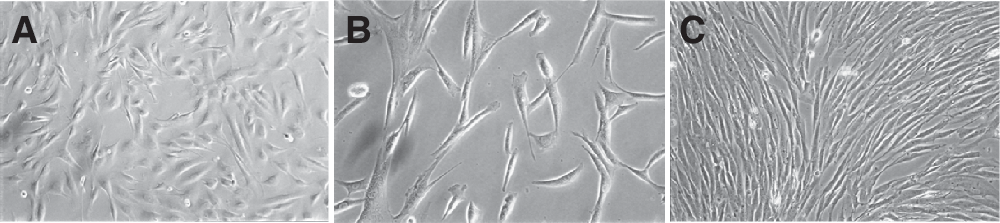
Morphology of nasal mucosa-derived mesenchymal stromal cells (nm-MSC). Standard phase contrast microscopy. Original magnification: 10× (
To analyze the clonogenic potential of these cells, CFU-F assays were performed from freshly isolated nasal mucosa-derived MSC. Whole cell preparations were plated and the frequency of colony-forming cells was determined on day 14. Five different donors were analyzed and the number of colonies was 154 ± 11.2/0.5 × 106 seeded cells. Large colonies consisted of >50 cells and the frequency of colony-forming cells in whole cell nasal mucosa preparations was calculated to be 0.03% (Fig. 2A and 2B). The cell doubling times (DT) across 10 passages (Fig. 2C) were monitored from three independent donors and showed a minor increase after passage 3 and a strong increase after passage 9. The overall mean DT across 10 passages was calculated as 2.42 ± 0.33 days/CD.
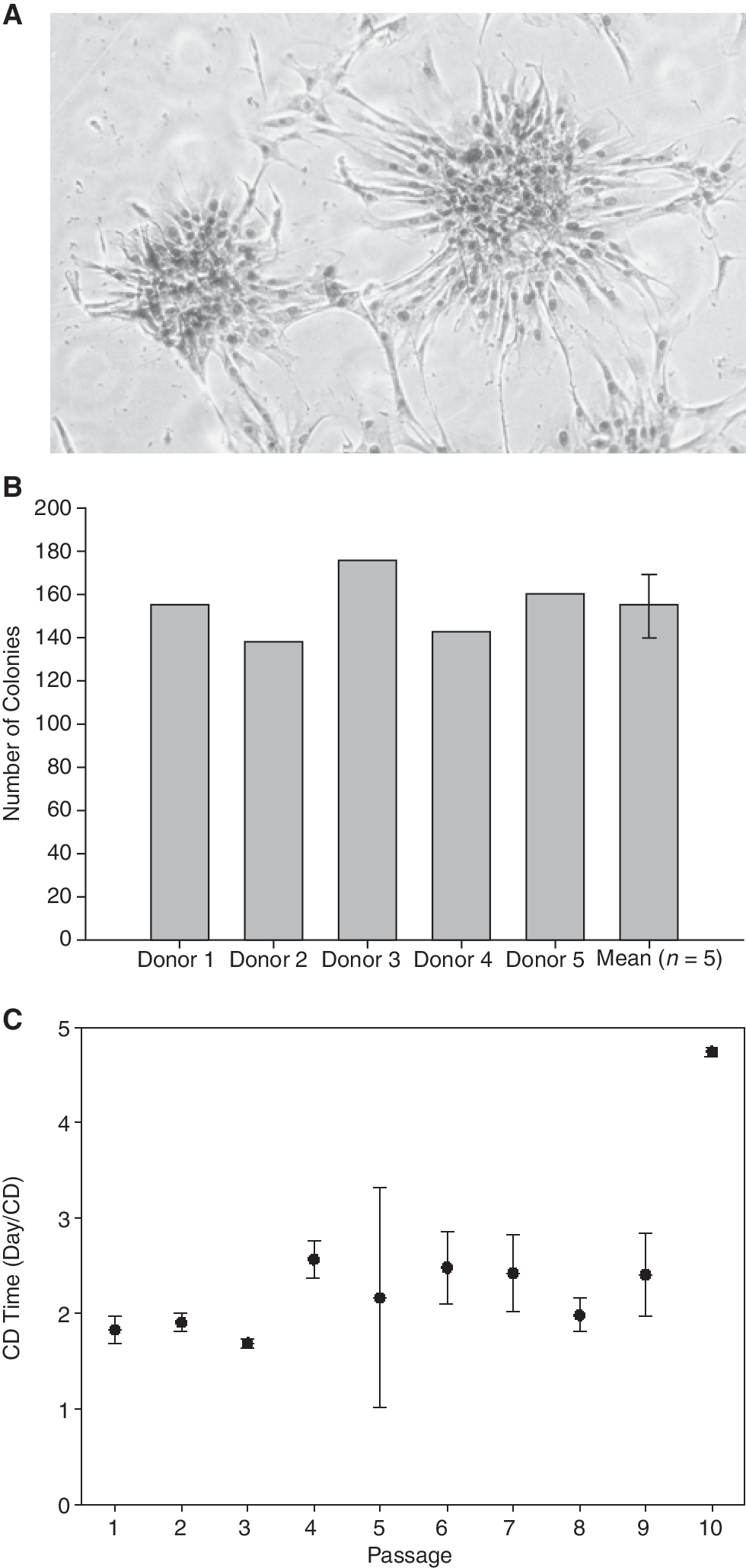
Standard colony-forming unit fibroblast assay (CFU-F). CFU-F assays were performed from primary cells of the nasal mucosa. The number of colonies was counted after 14 days of culture. (
Adherent colony-forming cells express surface markers of mesenchymal stromal cells
Flow cytometry analysis using multiple surface antigens demonstrated that nasal mucosa-derived fibroblast-like cells expressed typical MSC marker proteins. Identical results were obtained with cells from passages 2 to 8. MSC were strongly positive for CD29, CD44, CD73, CD90, and CD105. These tissue-resident MSC reproducibly expressed low levels of CD54 and CD117 (Fig. 3).

Flow cytometry analysis of nasal mucosa-derived mesenchymal stromal cells (nm-MSC). Data are shown as an overlay histogram: isotype control (gray) and specific cell-surface markers (white). Representative results from 1 out of 5 independent experiments are shown. Identical results were obtained with cells from passages 2 to 8. Cells were labeled with antibodies against CD11b, CD14, CD19, CD29, CD31, CD34, CD44, CD45, CD54, CD71, CD73, CD90, CD105, CD106, CD117, CD271, and STRO-1.
In contrast, known hematopoietic stem cell (HSC), neural stem cell (NSC), and endothelial cell markers such as CD11b, CD14, CD31, CD34, CD45, CD71, and CD106 were not expressed (Fig. 3). The surface antigens STRO-1 and CD271 are characteristic for bone marrow stromal cell progenitors and were not expressed on nm-MSC.
Trilineage differentiation of human nasal mucosa-derived MSC
We assessed the differentiation potential of human nasal mucosa-derived MSC by using cells from the second to the eighth passages. To examine whether these cells were able to undergo adipogenic differentiation, 5 independent cell cultures were subjected to 3 cycles of cultivation in adipogenic induction and maintenance media. At the onset of differentiation, no adipogenic cells were detected. Preadipocytes with lipid droplets appeared after one cycle of treatment. The lipid vacuoles became larger after 2 to 3 cycles of treatment (Fig. 4). After 3 weeks of culture, cells were positive for Sudan III. Control cells maintained in regular non-inductive medium showed no evidence of small oil droplets in the cytoplasm. Randomly selected areas from treated plates showed that ∼85% of the cells differentiated into lipid-producing adipocytes. The ability of these cells to differentiate into adipocytes was observed in both early (second) and late (eighth) passage, which is similar to adipose-derived MSCs. RT-PCR also demonstrated gene expression of adipogenic marker genes aP2 and C/EBPα (Fig. 4).

Trilineage differentiation of nasal mucosa-derived mesenchymal stromal cells (nm-MSC). Please note:
To examine the ability of MSC to differentiate into chondrocytes, cells were induced to form spheres by centrifugation in a 15-mL polypropylene tube and culture in either standard medium or chondrogenic induction medium. The ability of nm-MSC to undergo chondrogenic differentiation was confirmed by immunofluorescence analysis of collagen type II expression and by RT-PCR analysis of collagen type II and aggrecan gene expression (Fig. 4). We also demonstrated chondrogenic differentiation by Toluidine blue and Alcian blue staining, which demonstrated the presence of acidic mucopolysaccharides after 4 weeks of culture in chondrogenic induction medium (data not shown). However, this was not observed when cells were cultured in control standard medium.
To examine whether MSC were capable of osteogenic differentiation, the nm-MSC were cultured in osteogenic induction medium for 2–3 weeks. At day 0, most of the cells were negative for osteogenic markers, although very few cells expressed alkaline phosphatase (AP). At day 10, the expression of osteogenic lineage markers was strongly increased. Subsequently, from day 15 these cells showed positivity for alkaline phosphatase (AP) activity, immunofluorescence staining (osteopontin), and Alizarin red S, while cultures in control medium were negative (Fig. 4). RT-PCR analysis of osteogenic gene expression showed an up-regulation of osteopontin and osteocalcin, confirming differentiation on the level of gene expression (Fig. 4). Potential for osteogenic differentiation was higher in early passages compared with later passages. However, MSC colonies maintained differentiation potential over several passages.
Nasal mucosa-derived MSC are immunologically active and responsive
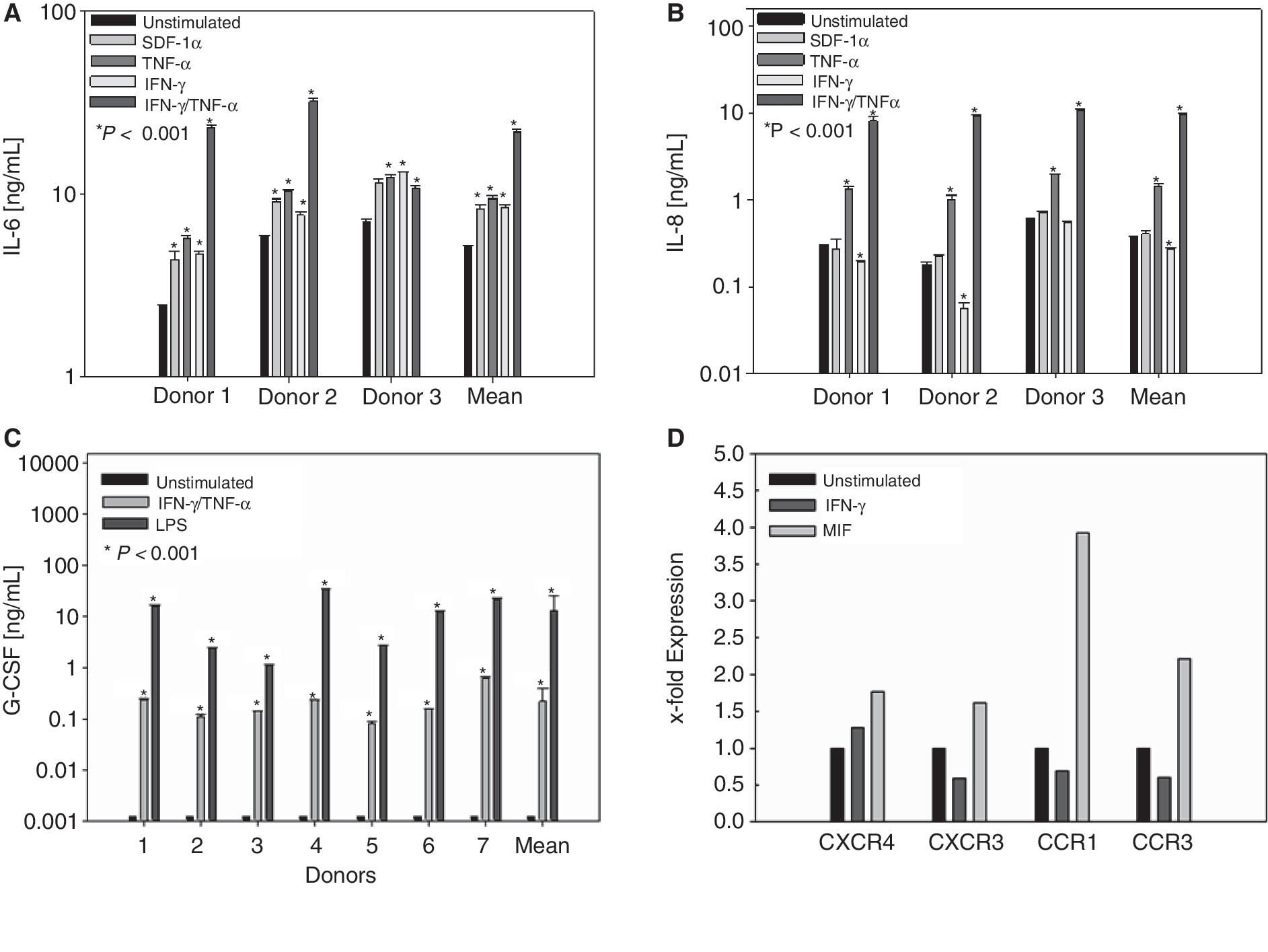
To test whether nm-MSC display immune cell-like features, secretion of cytokines, expression of chemokine receptors, and response to inflammatory signals were analyzed. Secretion of IL-6, IL-8, IL-10, TNF-α, IFN-γ, G-CSF, and MIF was determined by ELISA from third-passage MSC. nm-MSC secreted high level of inflammatory cytokines IL-6 and IL-8 and MIF that easily exceeded 1 ng/mL under standard culture conditions (Fig. 5). No constitutive secretion of IL-10, TNF-α, G-CSF, or IFN-γ was detected (Fig. 5). Next, we investigated the responsiveness of nm-MSC to immunological signals and exposed these cells to the cytokines IFN-γ and TNF-α and the chemokine SDF-1. All 3 signals strongly induced expression of IL-6 in nm-MSC. If exposed to a combination of IFN-γ and TNF-α, nm-MSC secreted large amounts of IL-6 reaching levels between 10 and 100 ng/mL (Fig. 6A). TNF-α also strongly induced expression of IL-8, while this cytokine could not be induced by IFN-γ and SDF-1α (Fig. 6B).
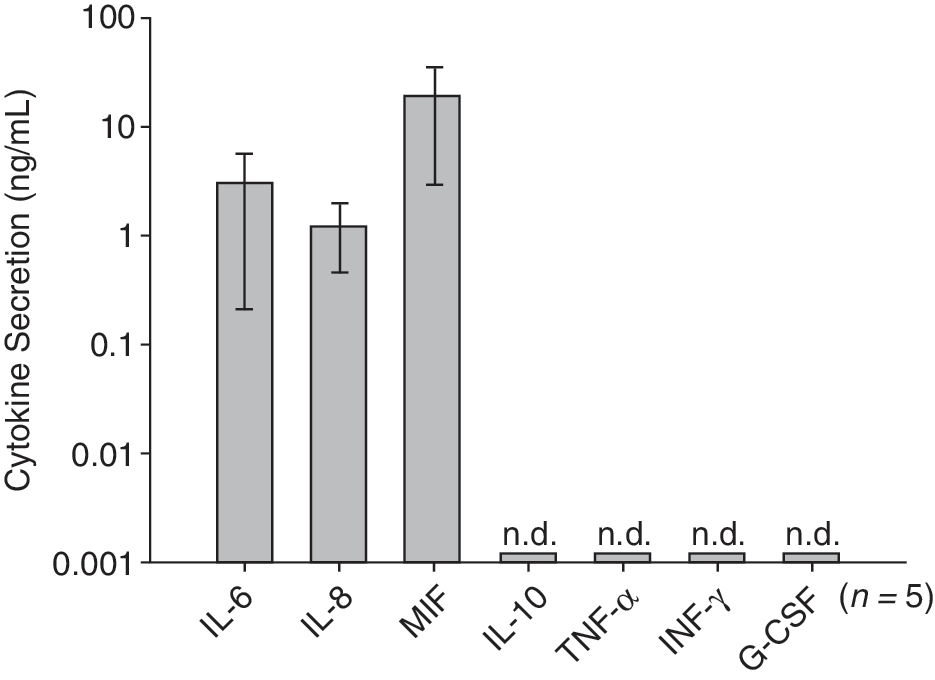
Constitutive cytokine secretion of nasal mucosa-derived mesenchymal stromal cells (nm-MSC). Third-passage stromal cells (5 donors) were washed 3 times with DMEM and cultured for 48 h in standard medium at the concentration of 1 × 106 cells/mL of medium. Secretion of IL-6, IL-8, IL-10, TNF-α, IFN-γ, G-CSF, and MIF was detected by enzyme-linked immunosorbent assay (ELISA). IL-10, TNF-α, IFN-γ, and G-CSF were not detectable (n.d.) in the nm-MSC supernatant.
(
If exposed to a combination of IFN-γ and TNF-α or LPS, nm-MSC secreted large amounts of G-CSF reaching levels between 0.8 and 10 ng/mL.
Finally, we analyzed expression of chemokine receptors of nm-MSC by RT-PCR. Unstimulated nm-MSC expressed CCR1, CCR3, CCR4, CCR6, CCR7, and CCR8, but were negative for surface expression of CCR2, CCR5, and CCR10. Other chemokine receptors that were expressed included CXCR1, CXCR3, CXCR4, CXCR5, CXCR6, CXCR7, CX3CR1, and XCR1. CXCR2 was not expressed (data not shown).
After we had demonstrated expression of a broad range of chemokine receptors, we analyzed regulation of these receptors by the Th1 cytokine IFN-γ and the inflammatory cytokine MIF. We observed that CXCR4 expression was up-regulated by both IFN-γ and MIF. MIF also induced expression of other chemokine receptors such as CXCR3, CCR1, and CCR3 (Fig. 6D). Interestingly, expression of these chemokine receptors was reduced after IFN-γ stimulation (Fig. 6).
Discussion
Bone marrow is the original and most prominent source of MSC. Consequently, bone marrow-derived MSC are well-characterized and the majority of studies in the field focus on this type of MSC [14]. Less is known about MSC from other sources, although considerable progress has been made during the last years to characterize MSC from other tissues besides bone marrow. Research in this field has been inspired by the fact that both bone marrow-derived MSC and tissue-resident MSC may have great potential for therapeutic application in cell-based medicine [15 –17].
Until now, there is no specific single marker to clearly define MSC. In fact, at present, a current definition for MSC as suggested by the International Society for Cellular Therapy (ISCT) uses a combination of morphological, phenotypical, and functional properties [2,3]. Clearly, MSCs and fibroblasts share a lot of functional and morphological properties such as phenotype, growth potential, differentiation potential, and immunoregulation. Therefore, common and distinctive features of both cell types are a matter of intensive debate [18 –20]. In the present study, we successfully isolated a novel population of mesenchymal progenitor cells from human nasal mucosa tissue. We use the term “mesenchymal stromal cells” (MSC), because we could demonstrate that nasal mucosa-derived MSC fulfill the minimal criteria for defining multipotent MSCs together with the ability to undergo adipogenic, osteogenic, and chondrogenic trilineage differentiation [3].
The morphological features of nm-MSC isolated with our method were similar to those described for bone marrow MSC, and included plastic adherence and fibroblast-like growth [21]. After an initial lag phase, the cells divided rapidly, with population doubling time depending on donor and initial plating density.
The classical assay used to identify colony-forming MSC and to determine their frequency is the colony-forming unit assay (CFU-U) that identifies adherent spindle-shaped cells that proliferate to form colonies [22]. We found a frequency of 0.03% MSC in nasal mucosal tissue. This represents a high frequency compared to other tissue sources like the bone marrow where the frequency is only 0.003% [23].
Several groups have examined the surface immunophenotype of MSC isolated from different tissues [14,24,25]. Immunophenotypic characterization of our isolated MSC demonstrated the presence of markers described for bone marrow-derived MSC (CD29, CD44, CD54, CD73, CD90, CD105) and the absence of hematopoietic (CD14, CD34, CD45) and endothelial (CD31) markers [2]. MSC are a well-characterized population of progenitor cells defined by their ability to differentiate into the multiple mesenchymal lineages [3]. As shown by immunofluorescence, immunohistology, and gene expression analysis of lineage-specific marker genes, nm-MSC shared the most important characteristic of MSC, namely, the capacity to differentiate into mesenchymal lineage (osteogenic, chondrogenic, adipogenic). These findings demonstrate that our isolation procedure effectively yields the generation of MSCs from nasal mucosa.
An emerging body of evidence shows that MSC possess immunomodulatory properties both in vitro and in vivo [26,27]. Earlier studies reported on the property of bone marrow MSC to inhibit proliferation and activity of T cells [28]. More recent studies have shown that MSC may down-regulate the function of different leukocyte subsets including dendritic cells (DC), B cells, and NK cells [29 –31]. Thus, bone marrow-derived MSC are currently regarded as a non-hematopoietic immunosuppressive cell type that inhibits effector functions of many immune cells. This feature is currently being therapeutically explored in settings of graft-versus-host disease (GvHD) after allogenic stem cell transplantation [32]. Systemic infusion of ex vivo-expanded MSC derived from adipose tissue was able to control lethal GvHD in mice transplanted with hematopoietic stem cells grafts [33].
Our study is the first to demonstrate isolation of MSC from mucosal tissue of the upper respiratory tract. As MSC interact with other immune cells, MSC may represent a thus far underestimated player in local immune regulation. To evaluate the potential role of nm-MSC in mucosal immunology, we tested key features of immunologic cell–cell interactions such as expression of different cytokines or chemokine receptors and the response of nm-MSC to selected cytokines and chemokines. Collectively, our experiments suggest that the nasal mucosa contains a population of MSC that is immunologically active and responsive.
nm-MSC constitutively secrete high amounts of IL-6, IL-8, and MIF, but not IL-10, TNF-α, and IFN-γ [34 –37]. Our experiments extend findings by others who found expression of IL-6 and IL-8 by bone marrow MSC together with a lack of IL-10 [4,38]. No constitutive secretion of G-CSF was detected. G-CSF is produced by endothelial cells, macrophages, and a number of other immune cells, and the G-CSF receptor is present on precursor cells in the bone marrow, and in response to stimulation by G-CSF, it initiates proliferation and differentiation of myeloid cells. G-CSF is also a potent inducer of HSCs mobilization from the bone marrow into the bloodstream, although it has been shown that it does not directly affect the hematopoietic progenitors that are mobilized [39]. After stimulation with LPS and TNF-α/IFN-γ, we detected strong secretion of G-CSF by nm-MSC.
Previously, expression of chemokine receptors by MSC has been reported [37,40]. We found expression of a wide range of chemokine receptors on nm MSC; some of which being shared with bone marrow-derived MSC [8]. Wynn et al. reported about high level of intracellular expression of CXCR4 [41]. In contrast, von Lüttichau et al. did not find an expression of CXCR4, but reported expression of CCR1, CCR4, CCR7, CXCR5, and CCR10, and these chemokine receptors were functional in driving MSC migration [42]. Thus, it appears that MSC express a variety of chemokine receptors, although there is much variability between different reports.
When we exposed nm-MSC to inflammatory mediators, we found that those mediators modulated the expression of chemokine receptors and the secretion of cytokines by nm-MSC. In particular, the combination of IFN-γ and TNF-α dramatically up-regulated inflammatory cytokine expression, while MIF induced expression of selected chemokine receptors.
In summary, our data show the presence of novel mesenchymal progenitor cell in the upper respiratory mucosal tissue. Based on the current understanding of MSC biology, those nasal mucosa-derived MSC may represent useful vehicles in cell-based medicine, tissue engineering, and regenerative medicine [43]. nm-MSC secreted classical immunoregulatory molecules and at the same time were also responsive to such signals. Therefore, our data suggest an important cross-talk between local immune cells and MSC in mucosal immunity. Future studies should address the role of MSC in mucosal immunoregulation in vivo to decipher a potential role of these non-hematopoietic cells in the course of local immune responses.
Footnotes
Acknowledgments
The authors would like to acknowledge Kirsten Bruderek, Petra Altenhoff, and Gisela Müller for excellent technical assistance, and Jan Hütte for help with preparation of figures. The authors thank Claudia Dumitru for critical reading of the manuscript. We are grateful to all patients and clinical colleagues, who donated or collected clinical samples. This project was supported in part by the Else-Kröner Fresenius Stiftung and the German Israeli Foundation.
Author Disclosure Statement
No competing financial interests exist.