
Abstract
The transient receptor potential melastatin type 7 channel (TRPM7) is a member of the TRP family of ion channels that is essential for cell proliferation and viability. Mesenchymal stem cells (MSCs) from bone marrow are a potential source for tissue repair due to their ability to differentiate into specialized cells. However, the role of TRPM7 in stem cells is unknown. In this study, we characterized TRPM7 in mouse MSCs using molecular biology, immunocytochemistry, and patch clamp. We also investigated TRPM7 function using a lentiviral vector and specific shRNA to knockdown gene expression. By RT-PCR and immunocytochemistry, we identified TRPM7, but not TRPM6, a close family member with similar function. Electrophysiological recordings during depletion of intracellular Mg2+ or Mg2+-ATP resulted in the development of currents typical for the channel. Furthermore, 2-aminoethoxydiphenyl borate (1 pM–100 μM) inhibited TRPM7 in a concentration-dependent manner. The molecular suppression of TRPM7 significantly decreased MSC proliferation and viability as determined by MTT assay. In addition, TRPM7 gene expression was up-regulated during osteogenesis. These findings demonstrate that TRPM7 is required for MSC survival and perhaps involved in the differentiation process.
Introduction
S
The transient receptor potential melastatin type 7 protein (TRPM7) is a member of the TRP family of ion channels originally identified from the Drosophila melanogaster visual system. TRPs are ubiquitous cation channels that are divided into 7 subfamilies: Group 1 (TRPC, TRPV, TRPM, TRPN, and TRPA) and Group 2 (TRPP and TRPML) [12]. TRPM7 is a member of the melastatin subfamily and is expressed in virtually all mammalian cells. Electrophysiological recordings revealed TRPM7 currents characterized by a large outward current at positive potentials and a small inward current at more physiological, negative potentials [13]. Divalent ions, mainly Ca2+ and Mg2+, account for the inward currents, while the outward currents are due to monovalent ions. Depletion of intracellular Mg2+ or Mg2+-nucleotides (Mg2+-ATP) activates TRPM7 [13 –15]. In resting cells, physiological levels of these molecules strongly suppress channel activity, and only a small constitutive activity remains to maintain basal divalent ion entry. TRPM7 currents are also referred to as Mg2+-inhibited cation (MIC, [16]) or Mg2+-nucleotide-regulated metal ion (MagNuM, [13]) currents. Activation of muscarinic receptors via Gi proteins inhibits TRPM7 currents, whereas stimulation of β-adrenergic receptors coupled to Gs proteins increases channel activity [14]. TRPM7 activity is also increased under low extracellular pH (4.0) conditions [17]. The absence of TRPM7 in DT-40 B cells and mast cells causes growth arrest and cell death [18,19]. These effects were reversed by supplementation of extracellular Mg2+ to TRPM7-deficient cells [18]. In rat basophilic leukemia cells, TRPM7 is up-regulated during the G1 phase [20]. Functional studies in zebrafish TRPM7 knockouts resulted in severe growth retardation and gross alterations in skeletal development, embryonic melanophore, and touch-response defects [21]. Deletion of the TRPM7 gene disrupts embryonic development and thymopoiesis in the mouse [22]. In mature bone cells, TRPM7 knockdown inhibits osteoblast proliferation [23]. Vascular smooth muscle growth and Mg2+ homeostasis [24], as well as retinoblastoma cell proliferation, are dependent on TRPM7 [25]. Most recently, TRPM7 was reported to control cell migration, which is critical for tissue remodeling and regeneration [26].
Given the importance of TRPM7 for cellular function, we investigated for the first time its presence in MSCs. We probed for TRPM7 gene expression and protein in bone marrow-derived MSCs and performed a detailed biophysical characterization of the channel. We also inhibited TRPM7 using a molecular approach to investigate its role in cell proliferation and viability.
Materials and Methods
Reagents
All reagents were purchased from Sigma Chemical Co. (St. Louis, MO), except 2-aminoethoxydiphenyl borate (2-APB), which was obtained from Cayman Chemical Co. (Ann Arbor, MI).
Cell culture
MSCs were isolated from male C57BL6 mice (25–30 g; Jackson Laboratory, Bar Harbor, ME) by flushing femur and tibia bones with cold IMDM medium containing 2% FBS, followed by filtration through a 70-μm mesh. Cells were grown in 100-mm round culture dishes and separated from hematopoietic stem cells based on their preferential attachment to bottom of the culture dishes for 2 passages. The presence of MSCs was confirmed by their ability to form fibroblast-like colony-forming units (CFU-F). Cells were grown in Mesencult™ medium (StemCell Technologies, Vancouver, BC, Canada) supplemented with 20% FBS and aerated with 5% CO2 and 95% air at 37°C. Experiments were performed with MSCs from passages 2 through 8. HEK 293 cells were cultured at 37°C with 5% CO2 in DMEM supplemented with 10% FBS.
Induction and detection of cell differentiation
Cells were cultured in growth medium supplemented with 10 nM dexamethasone, 0.1 mM ascorbic acid-2-phosphate, and 10 mM β-glycerophosphate. The medium was changed every 4 days during osteogenesis. After a 14-day period, the medium was aspirated and the cultures washed twice with PBS, fixed with 10% formaldehyde, washed again with distilled water, and incubated with 2% Alizarin Red S in distilled water (pH 4.1–4.3). After incubation, the staining solution was removed and the cultures washed with distilled water to get rid of excess dye. Stained monolayers were visualized by phase microscopy using an inverted microscope.
RT-PCR
Total RNA was extracted with RNAqueous-4PCR® kit according to the manufacturer's protocol (Ambion, Austin, TX). DNAse I-treated RNA was used for reverse transcription using RETROscript® Kit (Ambion). Equal amounts of RNA (1 μg) were used as templates in each reaction. PCR was performed by a standard method according to the manufacturer's protocol and results confirmed by the presence of specific bands visualized after agarose gel electrophoresis (1.5%), ethidium bromide staining, and UV transillumination. GAPDH primers were used as positive and ultra pure water as negative controls. The forward and reverse primer sequences were: TRPM7, CTCATGG GAGGAACCTACAG and CATCTTTGGTCTGTAGGGTTG (198 bp); TRPM6, GAAACTGTGGCTATTGGC and CCAATACGGCTCAAAGAC (317 bp); GAPDH, TGCTGAGTATGTCGTGGAGTCTA and AGTGGG AGTTGCT GTTGAA GTCG (650 bp).
Real-time RT-PCR (qPCR) was used to quantify TRPM7 mRNA levels in knockdown cells and during osteogenesis. A 7300 Real-Time PCR System and Taqman® primers and probes were used for qPCR reactions (Applied Biosystems, Foster City, CA). The reaction mixture contained 10 × Taqman® buffer, 5.5 mM MgCl2, 0.3 mM dATP, 0.3 mM dCTP, 0.3 mM dUTP, 0.3 mM dGTP, 900 nM forward primers, 900 nM reverse primers, 250 nM Taqman® probes, 0.3 U AmpliTaq® Gold DNA polymerase, 5 U MuLV reverse transcriptase, 7.5 U RNase inhibitor, and RNase-free water. Each sample (25 μL) was performed in duplicate. The qPCR conditions were 48°C for 30 min, 95°C for 10 min for one cycle, 95°C for 15 s, and 60°C for 1 min for 40 cycles. Results were expressed as the ratio of TRPM7 gene to that of the housekeeping gene, GAPDH.
Immunocytochemistry
Cells were grown on poly-
Electrophysiology
For patch clamp, cells were maintained in standard modified Ringer's solution of the following composition (in mM): NaCl 140, KCl 2.8, CaCl2 1, MgCl2 2, glucose 10, HEPES-NaOH 10, pH 7.2 adjusted with NaOH. The standard internal solution (SIS) contained (in mM): Cs-glutamate 140, NaCl 8, Cs-BAPTA 10, HEPES-CsOH 10, pH 7.2 adjusted with CsOH. The osmolarity of the solutions were ∼300 mOsm/L. The protocol for preparation of intracellular buffered Mg2+ and Mg2+-nucleotide concentrations was in general according to published methods (ref. [15] and Table 1).The concentrations were calculated with WebMaxC standard (
TRPM7 currents were recorded in the tight-seal whole-cell configuration mode at 21°C–25°C. High-resolution current recordings were acquired by a computer-based patch clamp amplifier system (EPC-10, HEKA, Lambrecht, Germany). Patch pipettes had resistances between 3 and 5 MΩ after filling with the SIS. Immediately following establishment of the whole-cell configuration, voltage ramps of 50 ms duration spanning the voltage range of −100 to +100 mV were delivered from a holding potential of 0 mV at a rate of 0.5 Hz over a period of 600 s. All voltages were corrected for a liquid junction potential of 10 mV between external and internal solutions, calculated using Igor PPT Liquid Junction Potential software (Wavemetrics, Portland, OR). Currents were filtered at 2.9 kHz and digitized at 100 μs intervals. Capacitive currents and series resistance were determined and corrected before each voltage ramp using the automatic capacitance compensation of the EPC-10. Since the divalent inward currents of TRPM7 are very small, the low-resolution temporal development of membrane currents was assessed by extracting the monovalent outward current amplitude at +80 mV from individual ramp current records.
Generation of TRPM7 knockdown clones and cell viability assay
Lentivirus plasmids were obtained from Sigma-Aldrich (St. Louis, MO) in a pLKO.1 backbone and contained either nonspecific control (SHC002) or shRNA specific for mouse TRPM7 (TRCN0000023955) under the control of the U6 promoter, plus the puromycin resistance gene. Lentiviruses were produced by triple transfections into 293T cells using Ca2+ phosphate transfection [27] and supernatants harvested (48, 74, and 96 h) and filtered (0.45 μm membrane). Viral particles were concentrated ∼100× using LentiX-concentrator solution (Clontech, Mountain View, CA) according to manufacturer's protocols and titered using serial Lv dilutions to infect 293T cells. The titer was determined by qPCR using genomic DNA and lentiviral vector-specific primers. MSCs were transduced overnight at a multiplicity of infection of 40 (MOI = 40) using Viraductin reagent (Cellbiolabs, San Diego, CA), according to manufacturer's protocols. Cells were allowed to recover from infection and 48 h later, puromycin added to the media at 1 μg/mL. The decrease in TRPM7 mRNA was confirmed by qPCR.
Stably transduced cells were used for MTT assay to compare the kinetic survival between control and TRPM7 knockdown groups. An equal number of cells (104/100 μL/well) were seeded into three 96-well plates 1 week after lentiviral infection and cultured in expansion medium. At different time points (36, 60, and 104 h), viable cells were examined by a MTT-based Cell Viability Assay Kit (Bioassay Systems, Hayward, CA). In brief, MTT solution (15 μL) was added to each well and incubated at 37°C for 4 h, and then the solubilization buffer (100 μL) was added to dissolve the insoluble formazan product. The absorbance of each well was measured at 550 nm using an Ultramark Microplate Imaging System (BioRad, Hercules, CA). Cell viability was expressed as the absorbance of the cultures subtracted from the background.
Data analysis
Patch clamp recordings are shown as means + s.e.m., and were plotted using Igor Pro 5 software program (Wavemetrics, Portland, OR). The OD values from control and TRPM7 knockdown groups in the MTT assay are shown as means ± s.e.m., and were compared using a 2-tailed, unpaired Student's t-test for each time point. Statistical significance was established at P < 0.05.
Results
TRPM7 expression in mesenchymal stem cells
We characterized TRPM7 in MSCs passages 2 through 8 because of the cells' consistent morphology (undifferentiated) and colony-forming capability (Fig. 1A). Since stem cells typically lose their differentiation capability over time under in vitro conditions, we examined whether MSCs from the highest passage studied (P8) were still capable of differentiating into bone. After a 14-day period in osteoblast differentiation medium, we detected mineralization in the cellular matrix using Alizarin Red S staining (Fig. 1B). We then performed a RT-PCR analysis to examine TRPM7 gene expression. We also probed for TRPM6 expression, a closely related family member with similar function. We identified TRPM7 transcripts with the expected molecular size of 198 bp, but not TRPM6 (317 bp) (Fig. 2A). Furthermore, the presence of the protein in MSCs was confirmed by TRPM7 immunoreactivity in colony cells (red) along with F-actin filaments (green) (Fig. 2B).
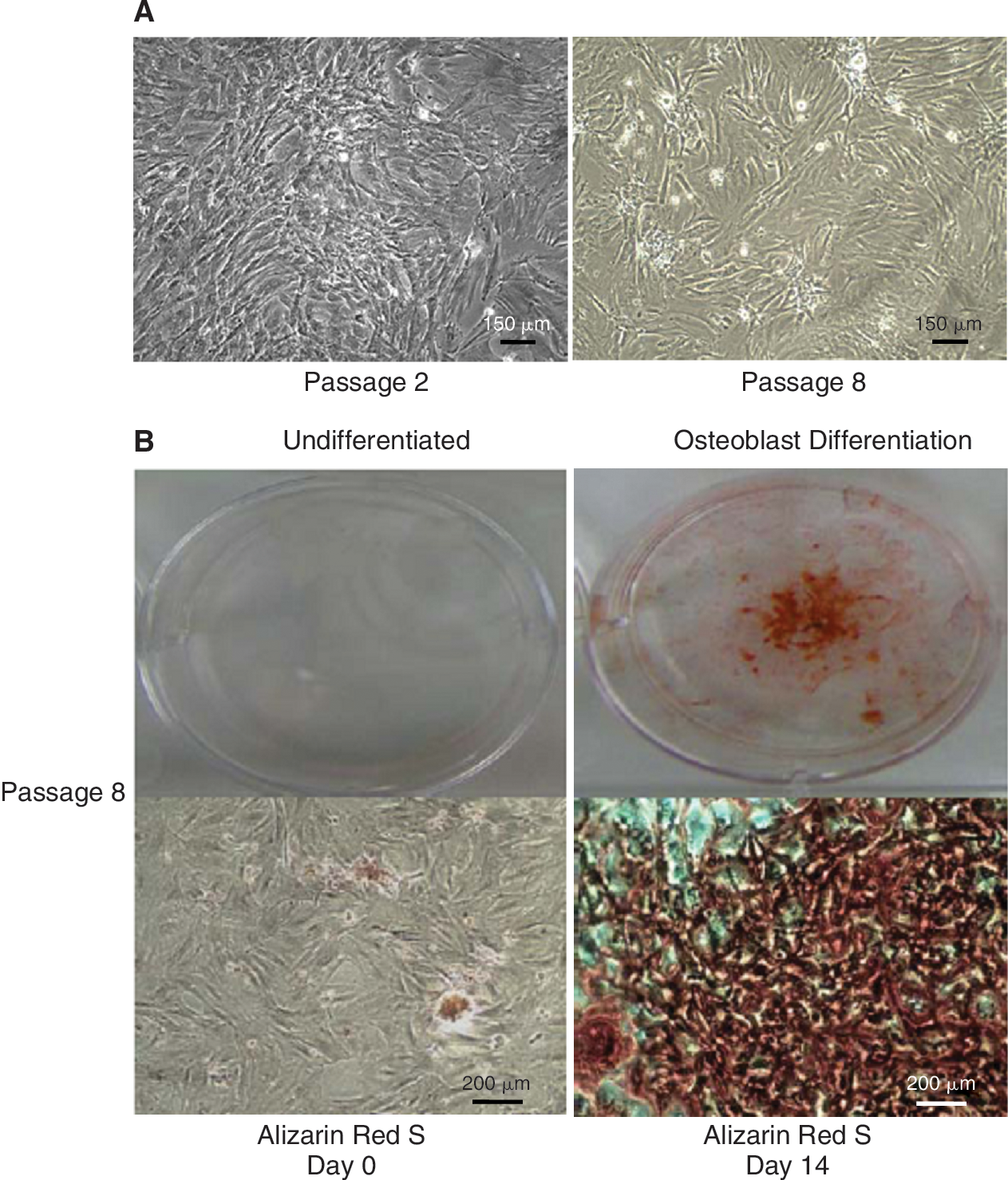
Morphology and differentiation capability of mesenchymal stem cells (MSCs). (
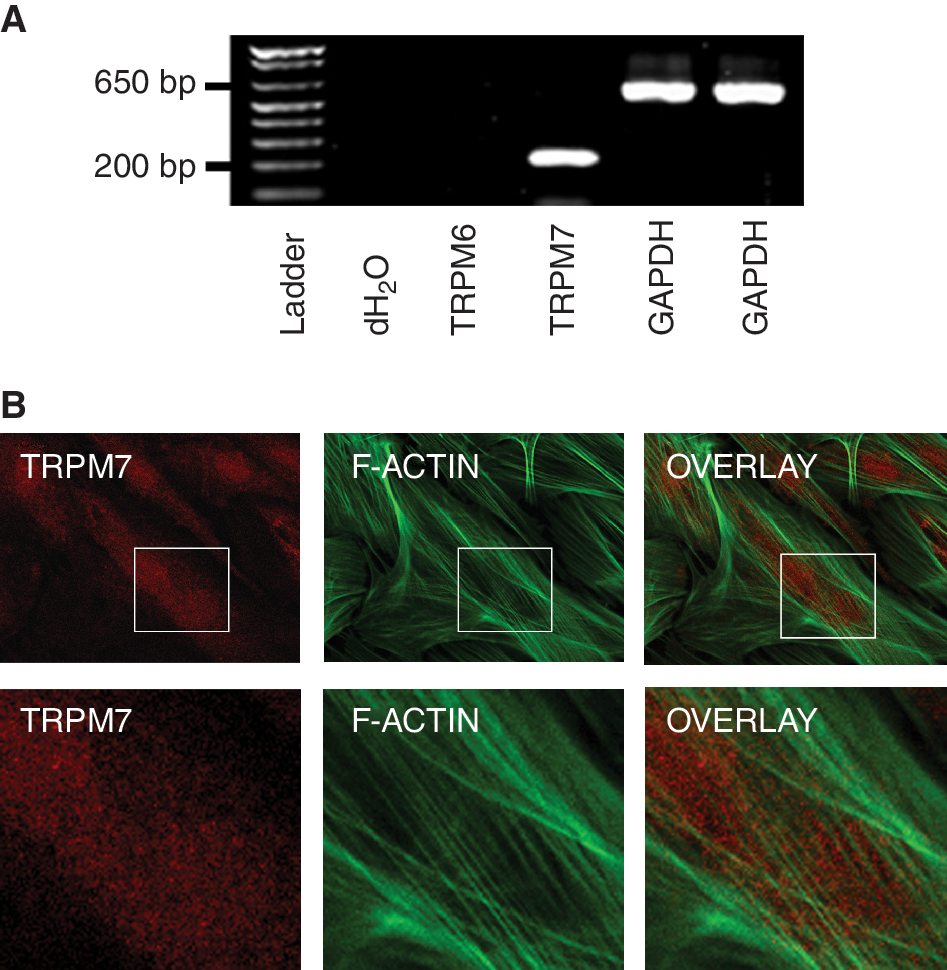
Expression of transient receptor potential melastatin type 7 channel (TRPM7) in mesenchymal stem cells. (
Endogenous TRPM7 currents in HEK 293 cells
When the intracellular concentration of Mg2+ or Mg2+-ATP decreases, TRPM7 channels are activated [13]. The currents generated by channel opening exhibit strong outward rectification, carrying monovalent ions outward at potentials above +50 mV and Ca2+ and Mg2+ into the cell at negative potentials. For experimental purposes, that is, very small divalent inward currents, we used the signature outward currents as a convenient measure of TRPM7 activity and demonstrated its characteristics in HEK 293 cells, which express endogenous channels. Depletion of intracellular Mg2+ from 3.19 to 0 mM activated TRPM7 in a dose-dependent manner (Fig. 3A). The maximum currents obtained under 0 mM Mg2+ conditions were in the range of 150 pA. The current-voltage (I/V) relationships taken from the peak current amplitude at 600 s for each Mg2+ concentration from representative cells resemble those described for TRPM7 (Fig. 3B). A dose–response analysis of the peak currents generated by depletion of intracellular Mg2+ resulted in a half maximal inhibitory concentration (IC50) and Hill coefficient of 0.525 mM and −1.5, respectively (Fig. 3C).
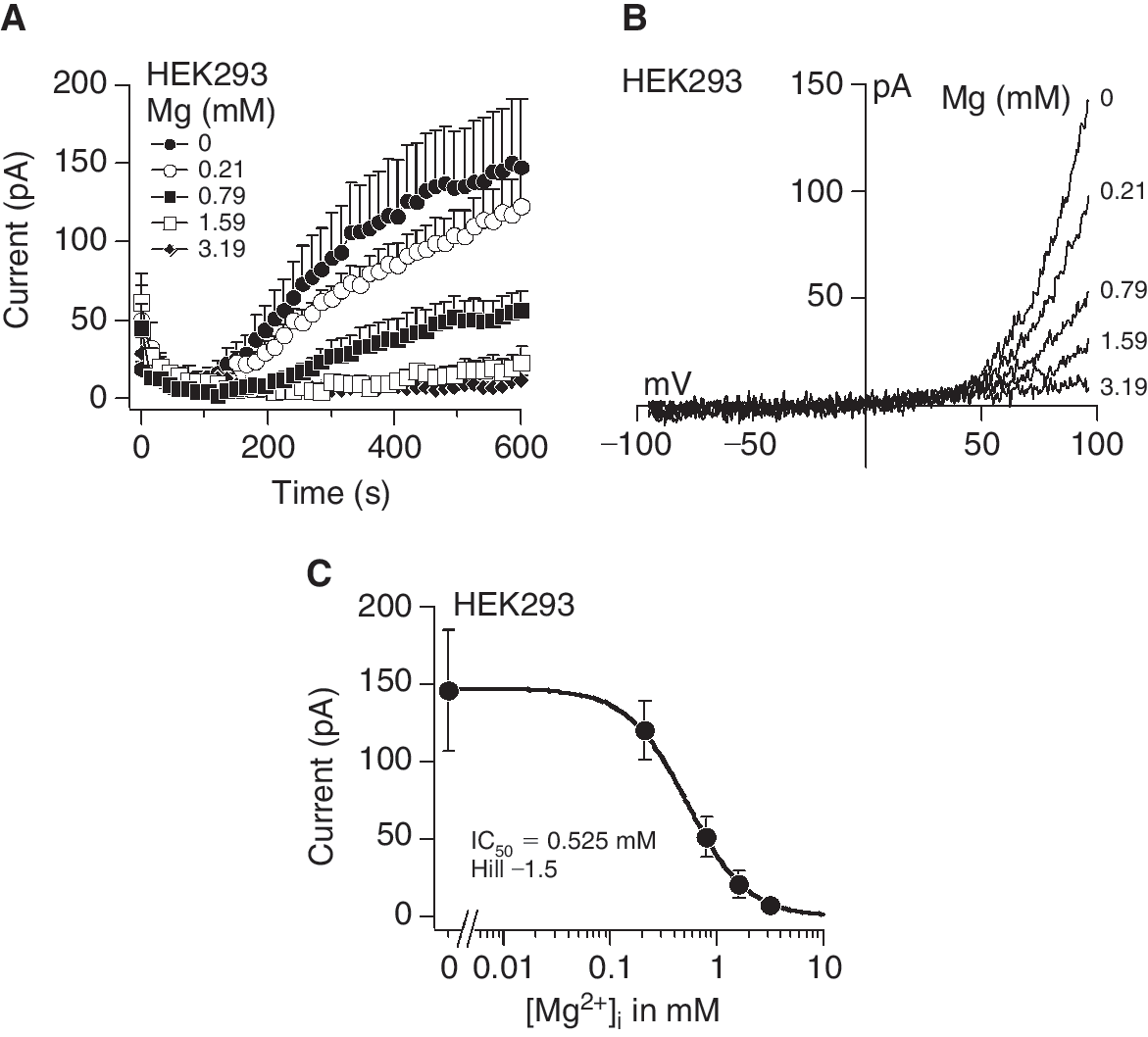
Activation of transient receptor potential melastatin type 7 channel (TRPM7) currents in HEK 293 cells. (
Characterization of endogenous TRPM7 in mesenchymal stem cells
Next, we examined whether TRPM7 currents could be detected in MSCs. A decrease in intracellular Mg2+ concentration from 1.59 to 0 mM activated TRPM7 in a dose-dependent manner (Fig. 4A). Perfusion of cells with 3.19 mM Mg2+ concentration inhibited TRPM7 similarly to 1.59 mM (data not shown). The maximum currents obtained under 0 mM Mg2+ conditions were similar to those in HEK 293 cells in the range of 150 pA and the I/V relationships taken at 600 s from representative cells were typical of TRPM7 (Fig. 4A and 4B). The calculated IC50 and Hill coefficient for Mg2+-induced inhibition were 0.130 mM and −1, respectively (Fig. 4C). In addition to Mg2+ alone, intracellular Mg2+-nucleotides (eg, Mg2+-ATP) can inhibit TRPM7 [15]. Since Mg2+ forms complexes with ATP (Mg2+-ATP) inside cells, we examined the interaction between both molecules by buffering the free Mg2+ concentration to 0.21 mM and varying the Mg2+-ATP concentration (Fig. 5 and Table 1).We selected 0.21 mM buffered Mg2+ because this concentration was in the range of the IC50 for Mg2+ inhibition and thus did not completely inhibit or activate TRPM7 (Fig. 4A and 4C). Increasing the intracellular Mg2+-ATP from 0 to 6 mM under 0.21 mM buffered Mg2+ concentration inhibited TRPM7 in a dose-dependent manner (Fig. 5A). Complete removal of ATP resulted in peak currents around 50 pA (Fig. 5A) and the I/V relationships taken at 600 s from representative cells are shown in Figure 5B. A dose–response analysis of the peak currents generated by decreasing Mg2+-ATP concentrations resulted in an IC50 and Hill coefficient for Mg2+-ATP of 2.7 mM and −2.3, respectively (Fig. 5C).

Activation of transient receptor potential melastatin type 7 channel (TRPM7) in response to depletion of intracellular Mg2+ in mesenchymal stem cells. (
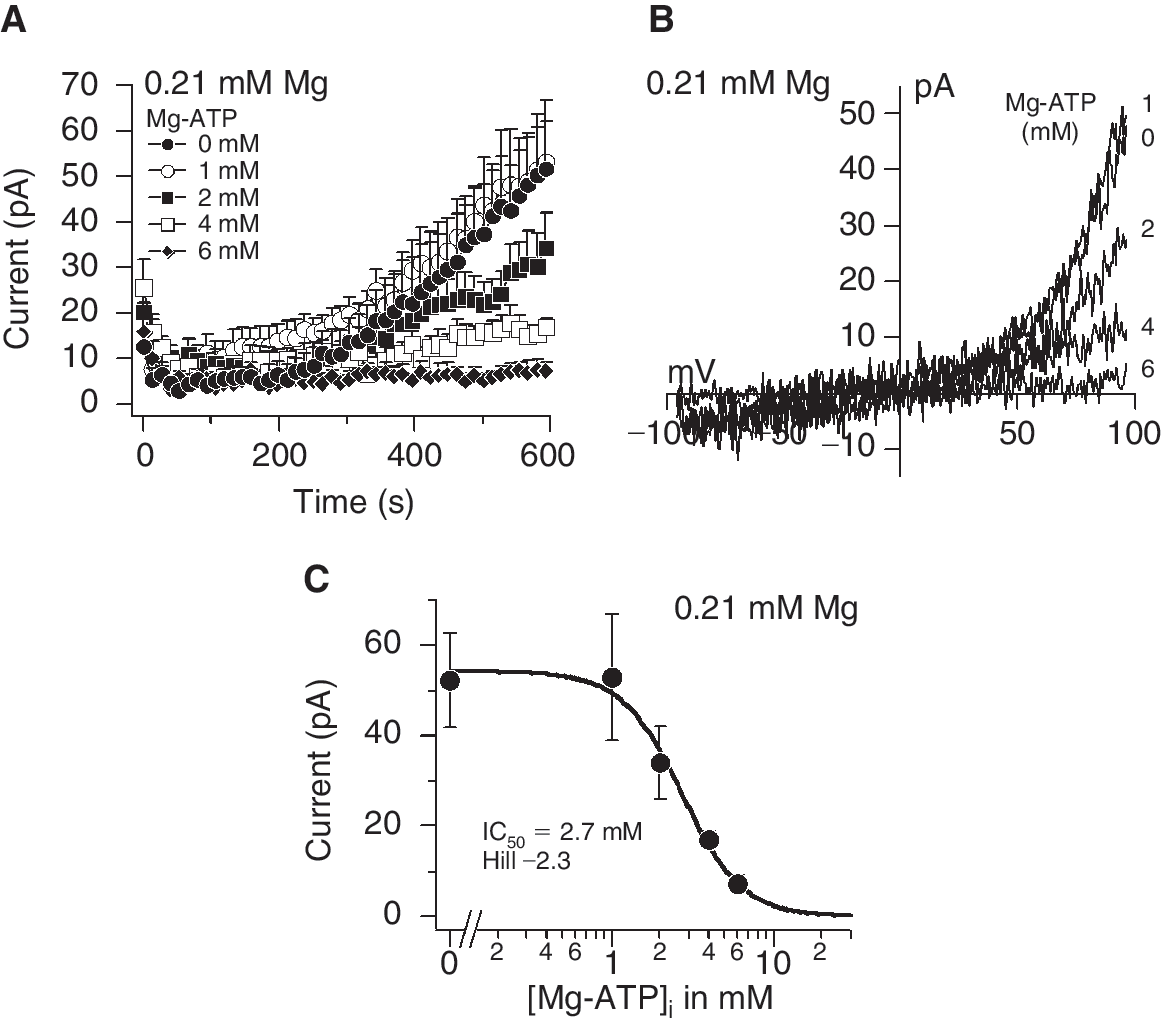
Inhibition of transient receptor potential melastatin type 7 channel (TRPM7) in response to intracellular Mg2+-ATP under buffered Mg2+ in mesenchymal stem cells. (
By increasing the concentration of internal Mg2+-ATP, the level of free ATP also rises (Table 1).To demonstrate that Mg2+-ATP (Fig. 5), but not free ATP inhibits TRPM7 currents in MSCs, we perfused the cells with 0 mM Mg2+ and 6 mM Na+-ATP. Figure 6A and 6B shows that 6 mM free internal ATP does not significantly affect the amplitude or the I/V relationship of 0 mM Mg2+-induced TRPM7 current.

Free ATP does not affect transient receptor potential melastatin type 7 channel (TRPM7) in mesenchymal stem cells. (
Inhibition of TRPM7 by 2-aminoethoxydiphenyl borate
Inhibition of TRPM7 by the compound 2-APB was reported in different cell types [28 –30]. Therefore, we utilized a pharmacological approach to confirm TRPM7 in MSCs. Pretreatment of cells with 2-APB 1 pM–100 μM inhibited TRPM7 currents generated by 0 mM Mg2+ in a dose-dependent manner (Fig. 7A). Exposure to 1 pM 2-APB resulted in TRPM7 currents similar to control cells treated with DMSO alone, whereas 100 μM completely blocked channel activity (Fig. 7A and 7C). The I/V relationships taken from representative cells at 600 s into the experiments resembles those of TRPM7 (Fig. 7B). A dose–response analysis of the peak currents with different 2-APB concentration resulted in an IC50 of 22.4 nM and a Hill coefficient of −0.33 (Fig. 7C).
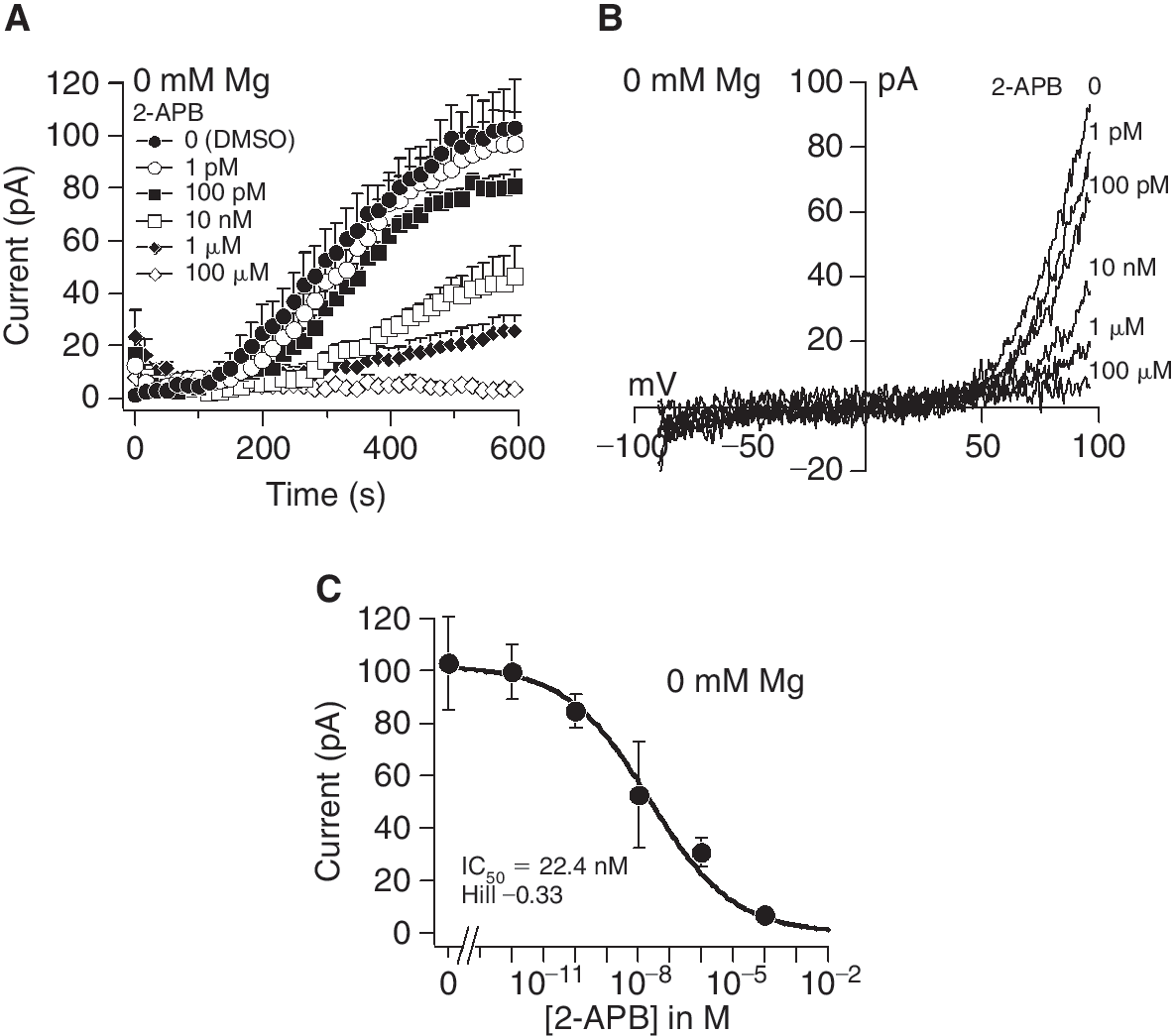
Inhibition of transient receptor potential melastatin type 7 channel (TRPM7) by 2-aminoethoxydiphenyl borate (2-APB) in mesenchymal stem cells. (
TRPM7 knockdown induces mesenchymal stem cell death
Previous reports in mature osteoblasts and immune cells show that inhibition of TRPM7 decreases cell proliferation, causes growth arrest and death [18,19,23]. Having confirmed the presence of TRPM7 in MSCs, we examined its function during cell proliferation and viability. We selected a lentiviral vector and shRNA to generate a stable knockdown system over conventional siRNA because of concerns regarding loss of knockdown effect caused by cell division. A 3.6-fold decrease in TRPM7 mRNA was obtained in knockdown cells compared to nonspecific shRNA controls (Fig. 8A). The effect of TRPM7 suppression on cell growth was visible after 14 days in expansion medium (Fig. 8B). Inhibition of TRPM7 significantly reduced the survival rate of MSCs at 36, 60, and 104 h compared to control cells at the same time point (Fig. 8C).

Transient receptor potential melastatin type 7 channel (TRPM7) knockdown induces mesenchymal stem cell death. (
TRPM7 expression is up-regulated during osteogenesis
In another experiment, we determined whether TRPM7 expression changes during the course of cell differentiation. This could provide some evidence for its role in osteogenesis. Hence, we quantified TRPM7 mRNA levels at days 7 and 14 during osteoblast differentiation. Alizarin Red S staining could be detected as early as day 7 with significant dye accumulation at day 14 (Fig. 9A–9C). The qPCR analysis with RNA extracted from duplicate flasks revealed an increase in 1.3- and 1.8-fold in TRPM7 expression on days 7 and 14 compared to control day 0 (Fig. 9D).

Transient receptor potential melastatin type 7 channel (TRPM7) gene expression is up-regulated during osteoblast differentiation. (
Discussion
Stem cell therapy offers a promising approach for the treatment of human as well as animal diseases due to their ability to regenerate damaged tissues (eg, muscle, nerve, bone, and fat). In order for stem cells to differentiate, they must possess the ability to proliferate and migrate as well as be viable. However, the molecular signals controlling these processes remain largely unknown. In this study, we identified and characterized TRPM7 in bone marrow-derived MSCs. By RT-PCR, we identified TRPM7 transcripts with the predicted molecular size (198 bp), but not TRPM6. This is in agreement with previous reports that TRPM7 is more widely distributed than TRPM6. In addition to stem cells, it is present in the kidneys, heart, pituitary gland, bones, mast cells, embryos, and adipocytes. TRPM6 expression is confounded to the brush borders of the small intestine and the renal-convoluted tubule where it functions in Mg2+ absorption [31]. The presence of the TRPM7 protein could also be detected by immunocytochemistry. So far, only 2 ion channels are known to conduct Mg2+ into cells, TRPM6 and TRPM7, which suggests that in stem cells, TRPM7 may represent a main pathway controlling Ca2+ and Mg2+ homeostasis.
We identified TRPM7 gene expression and protein in MSCs; therefore, we performed a detailed biophysical characterization of the channel to determine whether it was functionally active. In the absence of intracellular Mg2+, the development of currents, kinetics, and I/V relationships obtained in MSCs were typical of those reported for TRPM7. Since internal Mg2+ inhibits TRPM7, its intracellular depletion resulted in a dose-dependent activation with peak currents around 150 pA under 0 mM Mg2+ conditions with an I/V relationship exhibiting strong outward rectification at potentials above +50 mV. The similarity in current amplitude between HEK 293 cells and MSCs suggests that they may have comparable number of endogenous channels. The calculated IC50 for free Mg2+ inhibition of TRPM7 in MSCs was 130 μM (Fig. 4C). This is lower than in HEK 293 cells (525 μM, Fig. 3C), which is closer to 700 μM in other cell types and overexpression systems [15,16,32]. A reasonable explanation for the lower IC50 for free Mg2+ in MSCs is that TRPM7 may have greater sensitivity to changes in intracellular Mg2+ concentrations compared to other cells.
In addition to free Mg2+ alone, Mg2+-nucleotide complexes are capable of regulating TRPM7 activity. However, the inhibition of TRPM7 by intracellular Mg2+-ATP is controversial. While some studies indicate that Mg2+ and Mg2+-ATP inhibits TRPM7 [13,15,18,25], others challenge this ability [33]. We examined in stem cells the co-regulation of free Mg2+ and Mg2+-ATP complexes in the presence of 211 μM Mg2+, a concentration close to the IC50 of Mg2+ inhibition in MSCs (Fig. 4C). The binding affinity of free Mg2+ with ATP makes it impossible to examine the effect of buffered Mg2+-ATP alone because of the equilibrium with free Mg2+ according to the dissociation constant. Perfusion of MSCs with increasing Mg2+-ATP concentrations (in the presence of 211 μM free Mg2+) resulted in a dose-dependent inhibition of TRPM7 suggesting a regulatory function of Mg2+-nucleotide on TRPM7 in stem cells. In the HEK 293 TRPM7 overexpression system, an IC50 of 3.3 mM Mg2+-ATP was obtained under 0.21 mM free Mg2+ conditions [15]. This is consistent with our results under the same free Mg2+ concentration in MSCs where an IC50 of 2.7 mM was obtained (Fig. 5C). Demeuse et al. [15] reported that not only Mg2+-ATP but also other tri- as well as diphosphate Mg2+-nucleotides are capable of enhancing the block of TRPM7 currents by free Mg2+ with different potency.
Since increasing the internal Mg2+-ATP concentration under 211 μM buffered free Mg2+ also increases the level of free ATP (Table 1), we demonstrated that the rising Mg2+-ATP levels (Fig. 5), but not the free ATP inhibited TRPM7 currents. Perfusion of the cells with 0 mM Mg2+ and 6 mM Na+-ATP, that is, 6 mM free internal ATP, does not significantly affect TRPM7 currents in MSCs, indicating that Mg2+-ATP and not free ATP mediates this inhibition. Contrarily, in cardiac myocytes, ATP seems to be necessary to prevent a rundown of TRPM7-like currents [34]. Here, phosphatidylinositol 4,5-bisphosphate (PIP2), probably generated by ATP-utilizing lipid kinases, is necessary for maintaining channel activity. This PIP2-dependent rundown of TRPM7 in cardiac myocytes started about 20 min after achieving whole-cell configuration. This difference is perhaps due to the amount of time of TRPM7 recordings in the present study (max. 600 s), in which no rundown was observed in MSCs. Furthermore, we confirmed TRPM7 in MSCs with a pharmacological approach. A dose ranging from 1 pM to 100 μM 2-APB resulted in a concentration-dependent inhibition of TRPM7 currents induced by 0 mM Mg2+ with a calculated IC50 of 22.4 nM. This value is much lower than an IC50 of 178 μM reported by Li et al. [28] in HEK 293 TRPM7 overexpressing cells and may reflect the cell type or an effect of 2-APB on other proteins, which could impact TRPM7 indirectly. The effect of higher 2-APB concentrations on different proteins (eg, store-operated channels, TRPs, and Ca2+ pumps) has been reported by others [35 –37]. Hence, the results obtained with 2-APB in our study clearly show a greater sensitivity for the blocker as indicated by current measurements.
Functional analysis of TRPM7 in MSCs revealed the importance of the channel for cell survival, since molecular inhibition significantly decreased cell proliferation and viability. It is well established that the Mg2+ content inside cells directly correlates with proliferation rates as it stimulates DNA and protein synthesis, whereas Mg2+ deprivation causes growth arrest [38,39]. Magnesium ions are an important cofactor of topoisomerase II, an enzyme involved in DNA replication, transcription, and repair [40]. Other studies show that Mg2+ is essential for G1 phase progression [20,41,42]. In vertebrate cells, TRPM7 functions as a major pathway for Mg2+ entry, and currently there are 2 known ion channels capable of conducting Mg2+: TRPM6 and TRPM7. In MSCs, it appears that TRPM7 is the main pathway for Mg2+ entry, since we could not detect TRPM6 gene expression in these cells. Most of the reports regarding TRPM7 function indicate that TRPM7 is necessary for cell proliferation and viability [13,18,19,23,43]. In our study, TRPM7 knockdown significantly increased MSC death, therefore, our findings are in agreement with those reported in other cell types. In contrast, a recent study by Inoue and Xiong [44] showed an increase in endothelial cell growth and proliferation after TRPM7 knockdown. This effect was due to enhanced activation of the ERK pathway, which is normally utilized by growth factors. Whether this is a compensatory mechanism or a particular feature of TRPM7 in endothelial cells remains to be determined.
An important observation was the increased TRPM7 gene expression during osteoblast differentiation. Severe growth retardation and gross alterations in skeletal development are present in zebrafish mutants whose phenotype results from a mutation in TRPM7 [21]. Deletion of the TRPM7 gene also disrupts embryonic development and thymopoiesis in the mouse [22]. These findings suggest a role for TRPM7 in differentiation and development. Although Mg2+ is essential for cell proliferation, the mechanism controlling Mg2+ homeostasis in stem cells remains largely unknown. Studies in fibroblast cells revealed that Mg2+ deficiency accelerates cellular senescence [45]. Since stem cells undergo numerous cycles of cell division while maintaining the undifferentiated state, Mg2+ might be of special importance to protect from early aging and keeping the toti- or pluripotency of these cells.
In conclusion, we report for the first time in MSCs the presence of TRPM7, a Ca2+ and Mg2+ conducting channel that is essential for cellular function. By RT-PCR and immunocytochemistry, we identified TRPM7 gene expression and protein. Electrophysiological recording revealed Mg2+-inhibited currents with the characteristics of those previously described for TRPM7, also regulated by Mg2+-ATP in synergy with free Mg2+. From a functional perspective, our data show that TRPM7 is critical for MSC survival because channel knockdown decreased cell proliferation and viability. The TRPM7 gene expression was also found to be up-regulated during the differentiation process. The characterization of TRPM7 in MSCs provides a unique opportunity to examine its role in stem cell biology.
Footnotes
Acknowledgments
We thank Dr. Shaomian Yao and Matthew Screpetis for assistance with MSC differentiation. This work was supported by a LSU-SVM CORP grant and in part from the National Institutes of Health: DE018871 to H.C.
Author Disclosure Statement
No competing financial interests exist.