
Abstract
Mammalian spermatogonial stem cells are a special type of adult stem cells because they can contribute to the next generation. Knockout studies have indicated a role for TRP53 and PTEN in insulating male germ cells from pluripotency, but the mechanism by which this is achieved is largely unknown. To get more insight in these processes, an RNAi experiment was performed on the mouse spermatogonial stem cell line GSDG1. Lipofectamine-mediated transfection of siRNAs directed against Trp53 and Pten resulted in decreased expression levels as determined by quantitative RT-PCR and immunoblotting. The effects of knockdown were examined by determining the expression levels of genes that are involved in reprogramming and pluripotency of cells, specifically Nanog, Eras, c-Myc, Klf4, Oct4, and Sox2. Additionally, the effects of TRP53 or PTEN knockdown on Plzf and Ddx4 expression were measured, which are highly expressed in spermatogonial stem cells and differentiating male germ cells, respectively. The main finding of this study is that knockdown of Trp53 and Pten independently resulted in significantly higher expression levels of the pluripotency-associated gene Nanog, and we hypothesize that TRP53 and PTEN mediated repression is important for the insulation of male germ cells from pluripotency.
Introduction
S
In contrast with SSCs, in vitro-cultured embryonic stem (ES) cells, derived from preimplantation embryos, are pluripotent and can give rise to all the various specialized cell types that make up the body. Subcutaneous injection of these cells into immunocompromised mice results in the formation of teratomas; tumors that consist of cell types from all 3 embryonic germ layers. After injection in blastocyst stage embryos, ES cells can participate in host embryonic development and contribute to the formation of all fetal cell types including germ cells. Interestingly, under in vitro culture conditions, mouse SSCs can spontaneously form ES-like colonies with enhanced expression of pluripotency genes and decreased expression of genes characteristic of SSCs. Pluripotency of these ES-like cells has been confirmed by their contribution to germ-line chimaeras following injection into blastocysts [3]. A subsequent study verified that a single lineage-specific SSC could give rise to pluripotent ES-like daughter cells and to colonies of SSCs [4]. This was confirmed by another study in which single germ cells that were sorted for the expression of OCT4 and the absence of C-KIT gave rise to pluripotent colonies [5]. After testis transplantation, ES-like cells had lost their former capacity to restore fertility in testis of infertile mice, but instead formed teratomas, reminiscent of true ES cells that are unable to restore fertility after transplantation [4,5]. Several recent reports have independently described that pluripotent stem cells can also be generated from biopsies of human testes [5 –8]. The mechanism by which mouse or human SSCs convert to pluripotency is unknown.
The capacity of SSCs to spontaneously convert from a unipotent to a pluripotent state implies that these cells provide a tool to study the mechanism behind pluripotency and nuclear reprogramming. Moreover, improving our understanding of the mechanism by which SSCs acquire pluripotency and how to induce this could provide new opportunities to obtain pluripotent cells from species such as cow and pig, in which attempts to derive ES cell lines have failed [9,10]. However, it is still unclear what causes the shift from unipotency to pluripotency and how to induce this transition. Interestingly, an increased susceptibility to testicular teratomas has been observed in mice deficient in transformation-related protein 53 (Trp53), that codes for a transcription factor and tumor suppressor that can arrest cells for repair of DNA damage [11]. SSCs isolated from neonatal and adult testis of Trp53 knockout mice convert more easily to ES-like cells [3,12]. Trp53 has also been shown to suppress the pluripotency-associated gene Nanog and thereby induces the differentiation of ES cells into cell types that undergo efficient Trp53-dependent cell cycle arrest and apoptosis [13]. Moreover, pluripotent embryonic germ cells are more easily established from primordial germ cells of Trp53-deficient embryos than from wild-type and heterozygous embryos [14]. Recent work demonstrates that loss of TRP53 can enhance the efficiency by which somatic cells are reprogrammed to a pluripotent state with defined factors [15 –19]. These results suggest an involvement of TRP53 in insulating germ cells, SSCs, and dedifferentiated somatic cells from pluripotency and that loss of this single gene can enhance the efficiency by which cells acquire pluripotency.
The association of pluripotency with oncogenic transformation is also seen with tumor suppressor and antagonist of the PI3K/AKT pathway phosphatase and tensin homolog (Pten), as loss of Pten leads to testicular teratoma [20]. Down-regulation of PTEN enhances the transformation of primordial germ cells into pluripotent embryonic germ cells and a similar phenotype is observed after hyperactivation of AKT, an effect that is suggested to be exerted by inactivation of Trp53 [14,21].
In the current study, the mechanism was investigated by which TRP53 and PTEN insulate SSCs from pluripotency. Therefore, expression levels of TRP53 and PTEN were down-regulated in in vitro-cultured SSCs and changes in expression of candidate target genes were examined. The effect of transient RNAi-mediated knockdown of TRP53 and PTEN was studied by measuring expression levels of factors that play a role in pluripotency of ES cells (Nanog [22,23], Eras [24]). Furthermore, the effect of TRP53 or PTEN knockdown was measured on Plzf expression, a spermatogonia-specific transcription factor required for stem cell maintenance [25,26] and on Ddx4 [DEAD (Asp-Glu-Ala-Asp) box polypeptide 4], which is expressed in primordial germ cells, spermatogonia, and at high levels in differentiating male germ cells of the adult testis [27,28]. Finally, we examined if c-Myc, Klf4, Oct4, and Sox2, which in conjunction can induce pluripotency in somatic cells [29 –33], were up- or down-regulated upon knockdown of TRP53 and PTEN. In this study, it is demonstrated that independent knockdown of Trp53 and Pten leads to a significant increase in the expression of the pluripotency marker Nanog. These results could indicate redundant roles for TRP53 and PTEN in the insulation of unipotent SSCs from pluripotency through suppression of Nanog expression.
Materials and Methods
Cell culture and transfection
The mouse SSC line GSDG1 [2] was obtained from the RIKEN BioResource Center Cell Bank in Japan. The cells were cultured under feeder-free conditions in 12-well plates coated with 1 μg/cm2 laminin (Sigma-Aldrich Chemie, Zwijndrecht, The Netherlands) as previously described [2,34]. Culture medium was supplemented with 0.1% penicillin/streptomycin (Invitrogen, Breda, The Netherlands) and 0.1% Fungizone (Invitrogen). Cells were routinely cultured from passage 16 to passage 44 by reseeding every 3 to 4 days at 1.0 × 105 cells per well. Mouse ES cells were a kind gift from Stieneke van den Brink of the Hubrecht Laboratory in Utrecht. ES cells were cultured under standard mouse ES cell culture conditions on mitomycin C-treated STO cells.
Stealth/siRNA duplex oligoribonucleotides (Invitrogen) were used to knock down the expression of Trp53 and Pten. The sense strand from these molecules is chemically modified so that it cannot enter the siRNA pathway. With just 1 active strand, off-target effects from Stealth oligos are minimized. Three different Stealth/siRNA duplexes against Trp53 and 3 against Pten were tested for their efficiency of knockdown. Quantitative Real-Time PCR (RT-PCR) for Trp53 and Pten expression resulted in the selection of one Trp53 siRNA (5′-GCCAAGUCU GUUAUGUGCACGUACU-3′) and one Pten siRNA (5′-GGAAAUCGAUAGCAUUUGCAGUAUA-3′) for the knockdown experiments. Universal Control siRNAs (Invitrogen) with GC contents similar to the specific oligos were used as negative controls. BLOCK-iT Fluorescent (FITC-labeled) oligos (Invitrogen) were used to assess transfection efficiency. One day before transfection, 1.2 × 105 cells were plated uniformly across the wells of laminin-coated 12-well plates. On the day of transfection, cells were at 40% to 50% confluence, allowing a long interval between transfection and final assay time, and minimizing the loss of cell viability due to cell overgrowth. All transfections were performed in triplicate with lipofectamine 2000 transfection reagent (Invitrogen) in final siRNA concentrations of 36 nM according to manufacturer's instructions. In brief, 2 μL lipofectamine per well was diluted in 48 μL OPTI-MEM-I reduced serum medium (Invitrogen) and incubated for 5 min at room temperature (RT). Next, 2 μL 20 μM siRNA, mock and fluorescent oligos were diluted in 48 μL medium each, added separately to 50 μL diluted lipofectamine, and incubated for 20 min at RT. The resulting volume of 0.1 mL/mL transfection solution/well was dispensed into the wells of the culture plates containing 1 mL supplemented [2,34] StemPro-34 SFM (Invitrogen) culture medium but without fetal calf serum and antibiotics. Cells were incubated at 37°C for 6 h, after which the transfection medium was replaced with 1 mL culture medium with fetal calf serum and antibiotics. Transfection efficiency was assessed visually by means of FITC-labeled negative control oligos and knockdown efficiency was measured quantitatively by RT-PCR. For fluorescence microscope analysis of FITC-labeled control transfected cells, cells were imaged using an Olympus IMT 2-F inverted microscope.
Quantitative RT-PCR
For quantitative RT-PCR, total RNA was isolated from samples with TRIzol Reagent (Invitrogen), according to manufacturer's instructions, and subsequently treated with RNase-Free DNase (Qiagen, Venlo, The Netherlands) to remove genomic DNA. Next, cDNA was prepared using SuperScript III Reverse Transcriptase (RT; Invitrogen) according to the manufacturer's instructions. In brief, every 40 μL of reaction mix contained 20 μL DNase-treated RNA, 150 ng Random Primers (Invitrogen), 8 μL 5× First Strand Buffer (Invitrogen), 2 μL 0.1 M DTT (Invitrogen), 2 μL 10 mM dNTPs, 0.2 μL RNase in Ribonuclease Inhibitor (Promega, Madison, WI), and RNAse/DNAse-free water was used to adjust the reaction volume. A cDNA synthesis reaction was also performed without RT to obtain -RT controls for the detection of potential genomic DNA contamination. Amplification and detection was performed with a MyIQ single-color real-time PCR detection system (Bio-Rad, Veenendaal, The Netherlands). PCR primer pairs (Isogen, Maarssen, The Netherlands) were designed using Beacon Designer 4 (PREMIER Biosoft International, Palo Alto, CA). Primer nucleotide sequences are provided in Table 1 and sequence analysis confirmed amplification of the genes of interest. Quantitative RT-PCR amplifications were performed with iQ-SYBR Green Supermix (Bio-Rad) and the optimal annealing temperatures of the primer pairs were determined experimentally with a temperature gradient. RT-PCR conditions were: 3 min at 95°C, followed by 40 cycles of 20 s at 95°C, 30 s at the specific annealing temperature (Table 1), and 10 s real-time detection at 80°C–82°C. Finally, a melt curve was generated. For each gene, a separate reaction was performed and standard curves of 10-fold dilutions (10 pg to 1 ag) supplemented each run, which were used for quantification with the relative standard curve method. The threshold cycle (Ct) was set within the exponential phase of the PCR and standard curve efficiencies were between 90% and 110%. After absolute quantification, expression levels were normalized to the housekeeping genes Rpl22 and Rpl27 [36], which showed stable expression between the samples as verified by geNorm analysis [37]. Relative gene expression levels between siRNA-transfected cells and mock-transfected cells were determined at 72 and 96 h after transfection and the expression levels are presented as fold increase compared to mock.
Primers for c-Myc, Klf4, Oct4, and Sox2 have been described previously [35].
Immunoblot analysis
TRP53 and PTEN protein knockdown in Trp53 and Pten siRNA and mock-transfected cells was tested at 3 time points, which were chosen based on protein half-life predictions (
Statistical analysis
Data is presented as mean ± s.e.m. of at least 3 independent experiments and the 2-tailed unpaired Student's t-test was used to determine the significance of differences. A P < 0.05 was considered to be statistically significant.
Results
Characterization of ES cells and SSCs
In routinely cultured SSCs and ES cells, expression levels were determined for factors related to pluripotency or SSCs by quantitative RT-PCR. ES cells showed distinctively higher expression levels of genes involved in pluripotency and reprogramming than SSCs (Fig. 1). Nanog and Eras expression levels were considerably higher in ES cells than in SSCs. Inverse patterns were observed for Plzf and Ddx4, which were expressed at significantly lower levels in ES cells than in SSCs. The reprogramming factors c-Myc, Klf4, Oct4, and Sox2, which can induce pluripotency in somatic cells [29 –33], were all expressed at higher levels in ES cells than in SSCs. Surprisingly, despite its alleged role in refraining SSCs from pluripotency, Trp53 expression levels were approximately equal in ES cells and SSCs. Pten expression levels were about 2-fold lower in ES cells than in SSCs (Fig. 1). It was presumed that RNAi-mediated knockdown of TRP53 or PTEN in SSCs would affect the mechanism by which these cells are insulated from pluripotency, and consequently, enhance the expression levels of pluripotency and reprogramming factors and decrease the levels of Plzf and Ddx4.
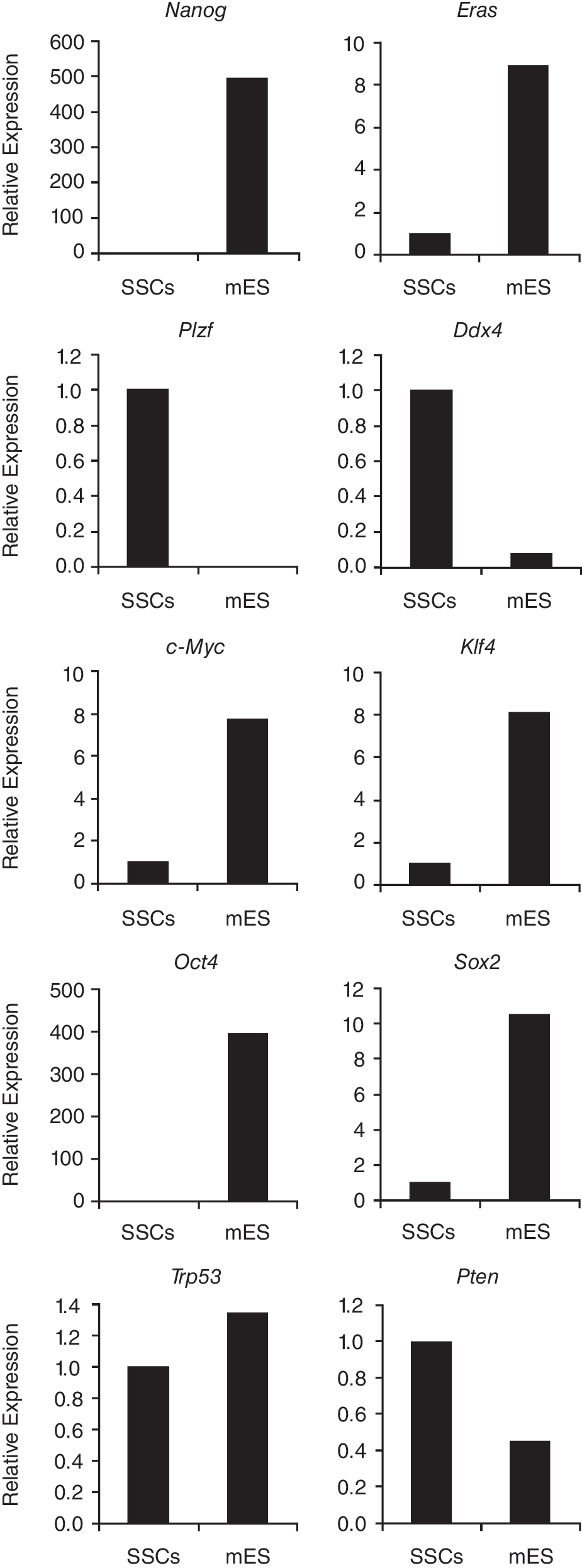
Relative mRNA expression levels, as determined by quantitative RT-PCR, in spermatogonial stem cells (SSCs) and embryonic stem (ES) cells of genes that are characteristic for pluripotent ES cells (Nanog and Eras), of SSC-marker Plzf and germ cell marker Ddx4, of genes that can reprogram somatic cells to a pluripotent state (c-Myc, Klf4, Oct4, Sox2), and of Trp53 and Pten. SSCs = SSC line GSDG1 [2], mES = mouse ES cells. Relative expression levels in SSCs were set to 1.
Efficiency of siRNA-mediated knockdown of Pten and Trp53
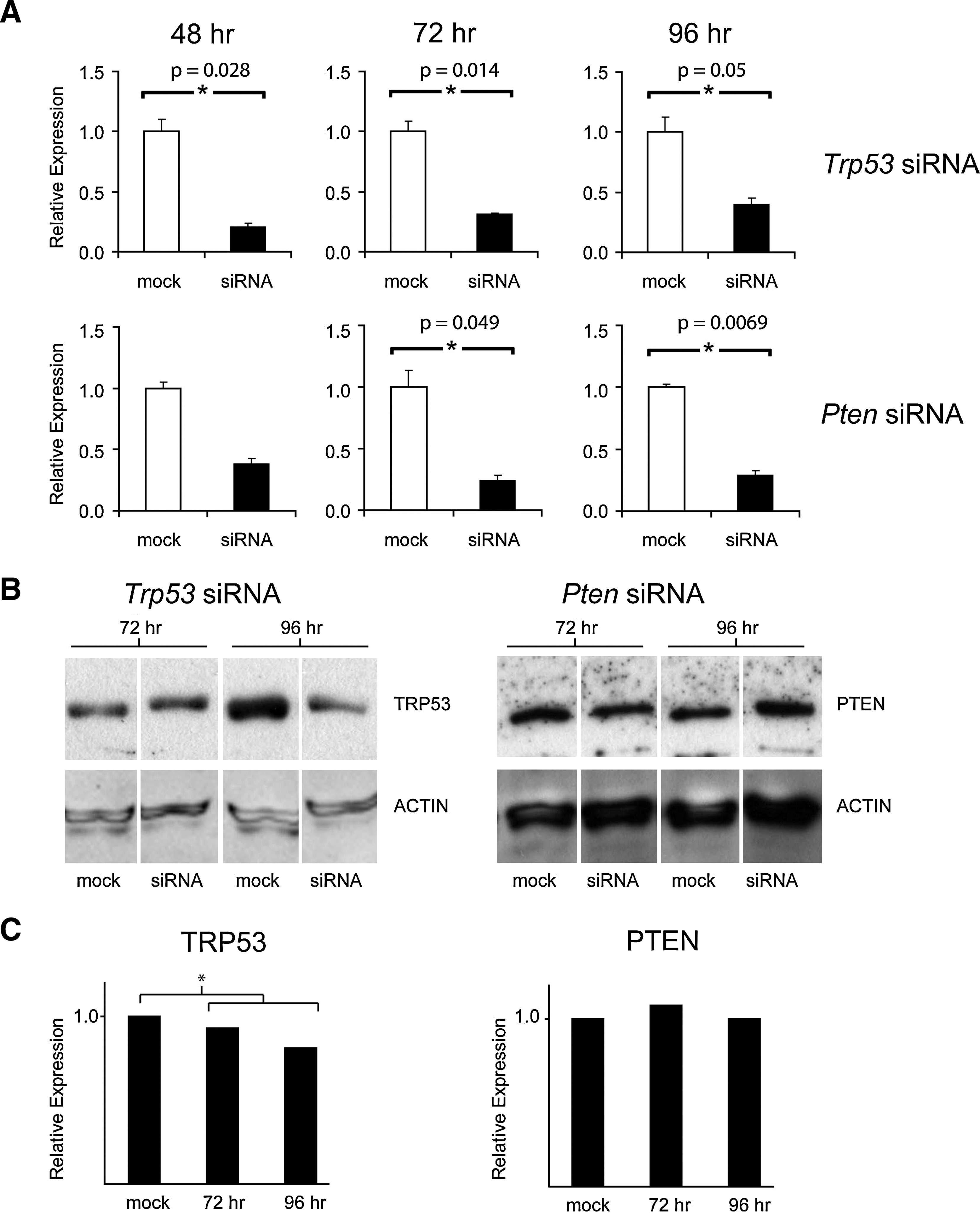
Transfection efficiency of SSCs was determined with FITC-labeled oligos. Eight hours after transfection, siRNA transfection efficiency was estimated between 95% and 100% and fluorescent signal could be detected up to 4 days after transfection (Fig. 2). Trp53 mRNA levels were significantly knocked down at 48, 72, and 96 h after transfection, as compared to mock-transfected controls (Fig. 3A) and the efficiency of Trp53 knockdown was 80%, 70%, and 60%, respectively. Moreover, knockdown of Trp53 resulted in a noticeable reduction of TRP53 protein levels in SSCs at 96 h after transfection, when compared to mock-transfected controls (Fig. 3B). A significant reduction in Pten mRNA levels of 75% and 70% was achieved at 72 and 96 h, respectively, after transfection with Pten-specific siRNAs (Fig. 3A). Despite this reduction in Pten mRNA expression levels, the anticipated decline in PTEN protein levels was not observed (Fig. 3B). Although PTEN protein levels appear to decrease at the 72-h time point, there was a high variability between biological replicates and significant differences could not be determined. Cell morphology was monitored before and after transfection, but no change in cell morphology was observed in siRNA-transfected cells when compared to untreated or mock-transfected cells.
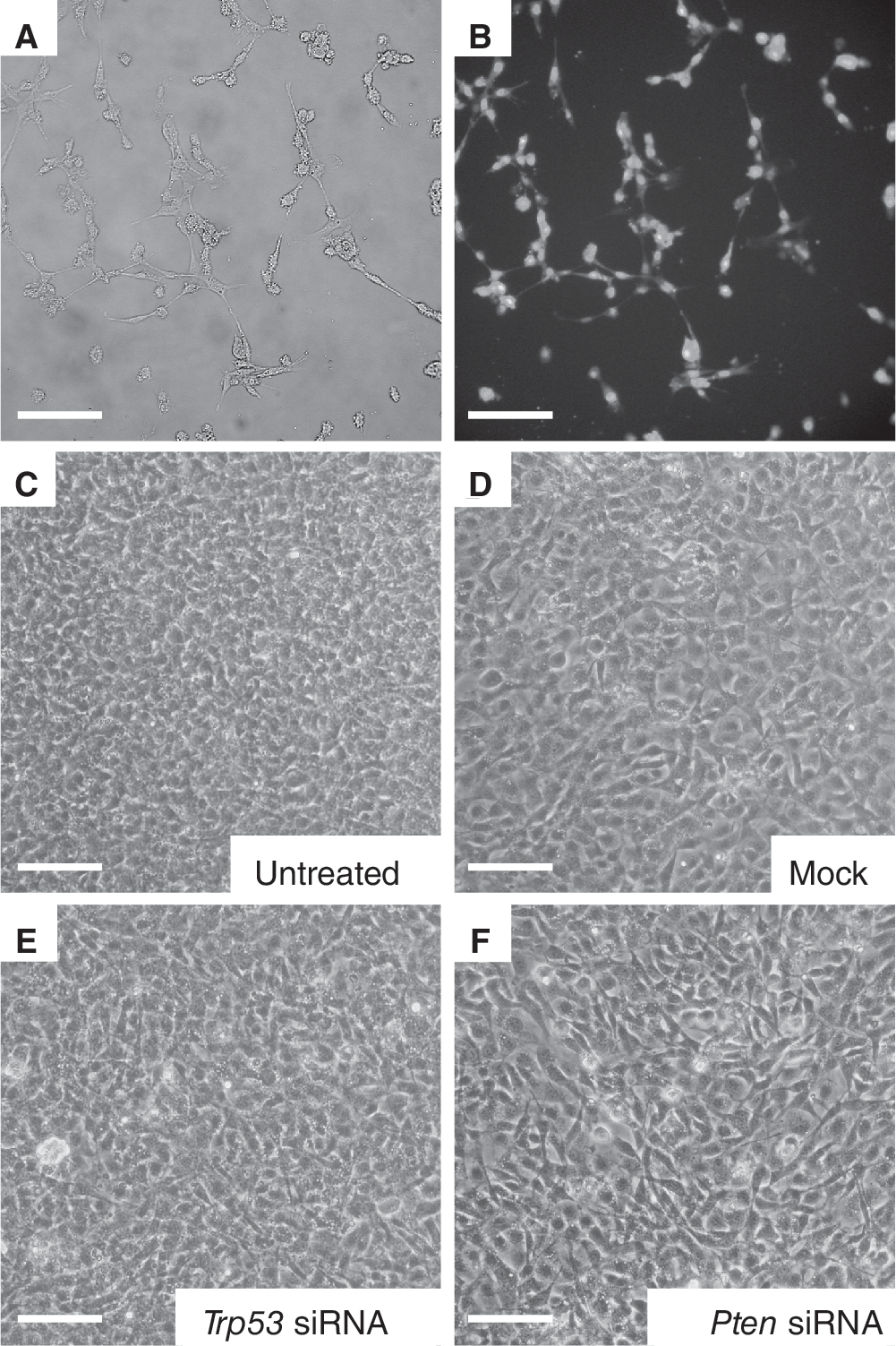
Transfection efficiency and cell morphology after knockdown of Trp53 and Pten. (
(
Effects of Trp53 and Pten knockdown on genes important for germ cells or pluripotency
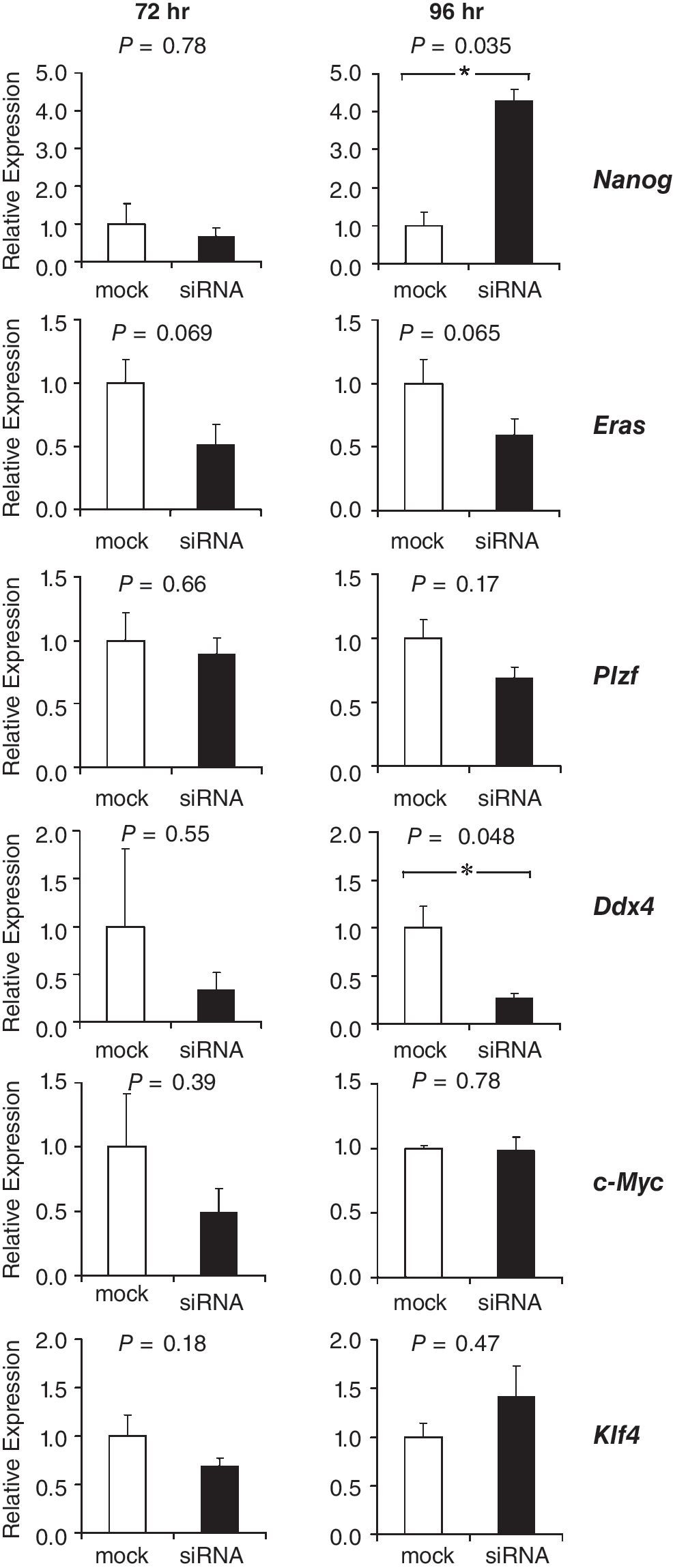
At 72 and 96 h after transfection, expression levels of pluripotency factors were measured to determine if TRP53 knockdown influenced the insulation of SSCs from pluripotency. Knockdown of TRP53 resulted in elevated levels of Nanog at 72 h after transfection and a ∼2-fold increase at 96 h after transfection, as assessed by RT-PCR (Fig. 4). Eras and Ddx4 expression levels were unaffected by TRP53 knockdown. Surprisingly, expression of the SSC factor Plzf significantly increased 2-fold at 96 h after transfection. At 72-h post-transfection, expression levels of c-Myc were slightly but significantly decreased, but at 96 h no differences were observed. Klf4 expression levels were unaffected by the knockdown. For most samples, expression levels of Sox2 and Oct4 did not reach the threshold cycles, which impeded a quantitative assessment of the effect of knockdown on the expression of these genes. Nevertheless, in 4 out of 6 samples in which TRP53 was down-regulated, Sox2 expression was detected. In comparison, just 1 out of 6 mock control samples expressed Sox2 (data not shown). TRP53 might therefore act as a repressor of Sox2 in SSCs.
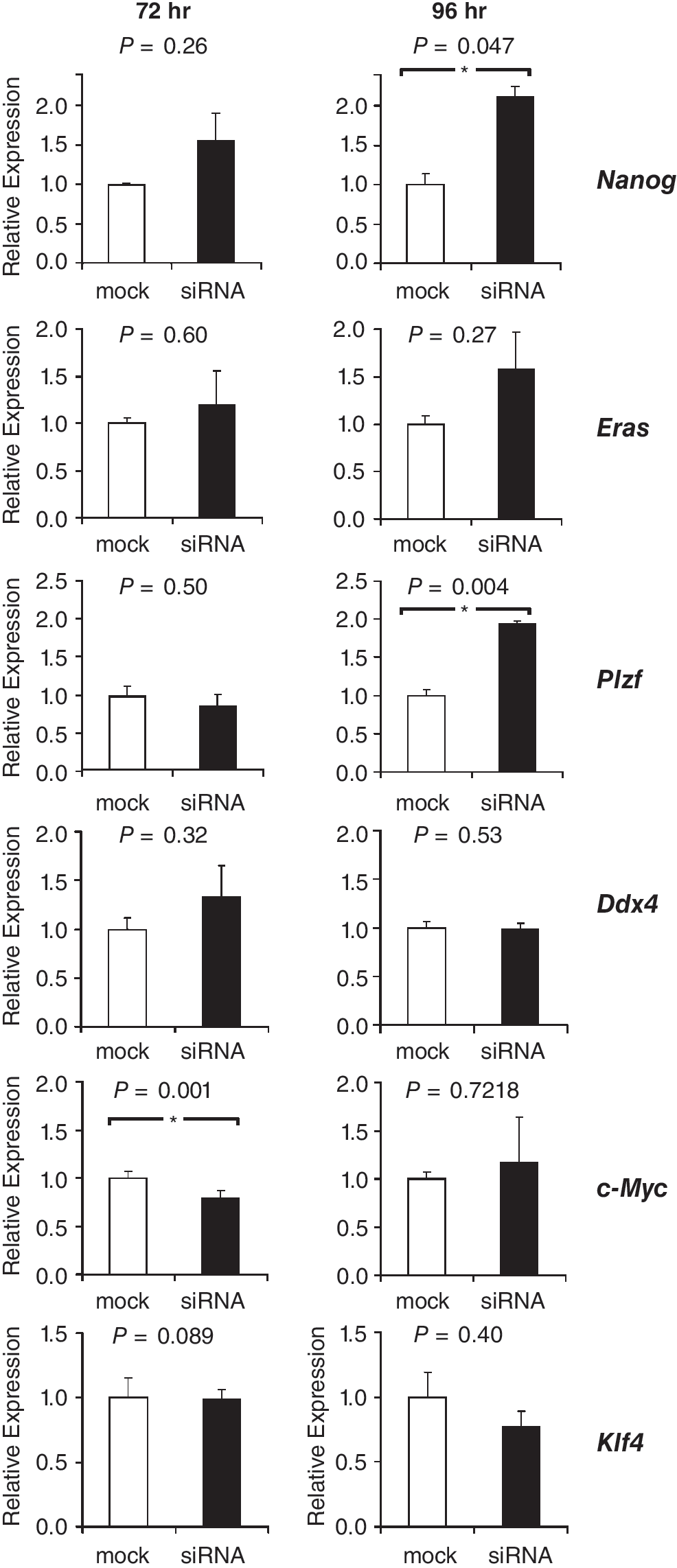
Relative mRNA expression levels of pluripotency genes (Nanog and Eras) of spermatogonial stem cell (SSC)-enriched genes Plzf and Ddx4 and of the genes encoding the reprogramming factors C-MYC and KLF4 at 72 and 96 h after transfection with siRNAs against Trp53. Expression levels in negative controls (mock) were set to 1. Levels are expressed as mean ± s.e.m. (n = 3). Asterisks denote significant differences in expression levels between negative controls (mock-transfected cells) and siRNA-transfected cells.
At 72 h after transfection with siRNA oligonucleotides against Pten, expression levels of Nanog were similar between siRNA-treated cells and mock-transfected controls (Fig. 5). However, at 96 h after transfection, Nanog was > 4 times up-regulated in cells in which PTEN was knocked down. Expression levels of pluripotency factor Eras and SSC factor Plzf were unaffected by the knockdown. However, at 96 h after transfection, Ddx4 was expressed at significantly lower levels in cells with decreased Pten levels. The expression levels of reprogramming factors c-Myc and Klf4 were not significantly altered upon knockdown of PTEN (Fig. 5). The expression levels of pluripotency factors Sox2 and Oct4 were below background levels and an effect of knockdown on the expression of these genes could not be determined (data not shown). Important for both experiments is that TRP53 knockdown did not influence Pten expression and, vice versa, PTEN knockdown did not influence Trp53 expression in SSCs (Fig. 6).
Relative mRNA expression levels of pluripotency genes (Nanog and Eras) of spermatogonial stem cell (SSC)-enriched genes Plzf and Ddx4 and of the genes encoding the reprogramming factors C-MYC and KLF4 at 72 and 96 h after transfection with siRNAs against Pten. Expression levels in negative controls (mock) were set to 1. Levels are expressed as mean ± s.e.m. (n = 3). Asterisks denote significant differences in expression levels between negative controls (mock-transfected cells) and siRNA-transfected cells.
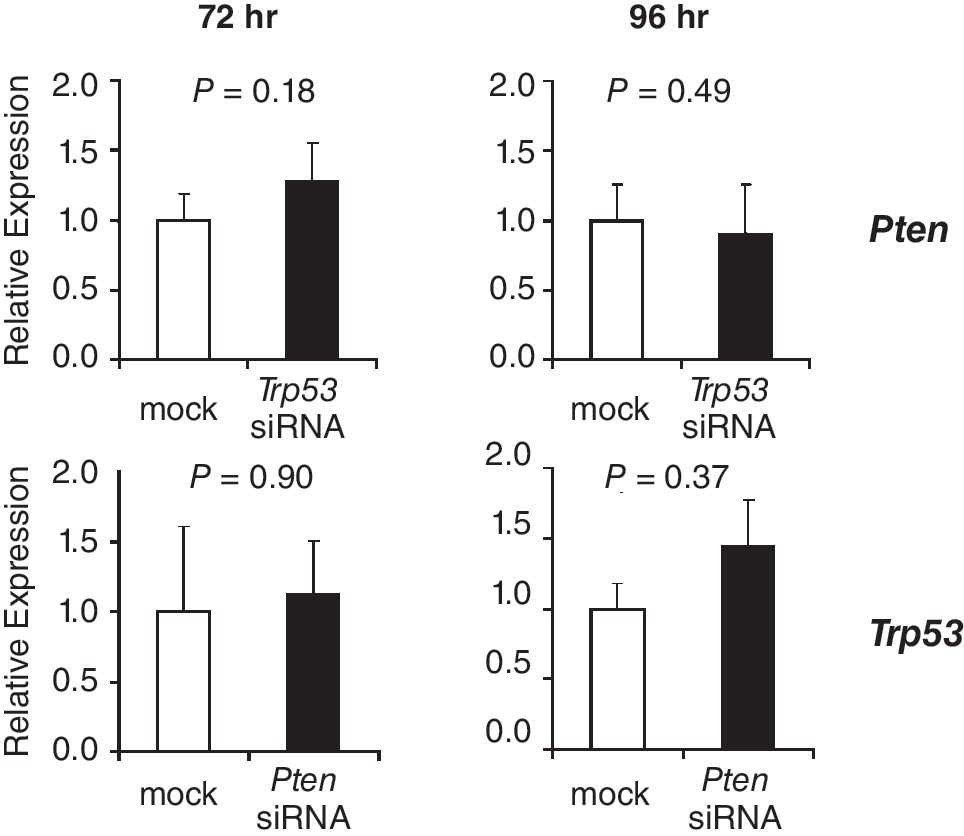
Relative mRNA expression levels of Pten and Trp53 at 72 and 96 h after transfection of spermatogonial stem cells (SSCs) with siRNAs against Trp53 and Pten, respectively. Expression levels in negative controls (mock) were set to 1. Levels are expressed as mean ± s.e.m. (n = 3).
Discussion
Under certain pathogenic or in vitro culture conditions, germ cells can acquire pluripotency as demonstrated by teratoma formation in gonads [39] and by the derivation of pluripotent cell lines from primordial germ cells and SSCs [3,40 –44]. Tumor suppressors TRP53 and PTEN and the PI3K-AKT signaling pathway have emerged as key players in refraining germ cells from pluripotency [3,12,14,20]. In the current study, the molecular mechanisms that trigger the conversion of male germ cells toward pluripotency were investigated by RNAi-mediated knockdown of Trp53 and Pten mRNA levels. TRP53 protein levels were significantly reduced by the mRNA knockdown, but a decline in PTEN protein levels could not be detected. PTEN, which has a similar calculated protein stability as TRP53 [38], showed an ∼20% less efficient mRNA knockdown at day 2 as compared to Trp53, which may have caused the lack of detectable protein knockdown. Lack of detection could also be caused by the limited resolution of immunoblot analysis. We have used Stealth siRNAs for the knockdown experiments, because the sense strands from these molecules are chemically modified so that they cannot enter the siRNA pathway. With just one active strand, off-target effects from Stealth oligos are minimized. Nevertheless, since PTEN protein levels were not noticeably affected by Pten siRNAs, the observed results of this siRNA treatment could be due to off-target effects of the oligo. Clear specific effects were observed at the gene expression level after the Pten siRNA treatment, as for the Trp53 siRNA treatment.
The findings of the current study suggest that TRP53 and PTEN suppress expression of the pluripotency factor NANOG in SSCs, because inhibition of Trp53 or Pten resulted in significantly elevated Nanog expression. It has previously been shown that PTEN protein antagonizes PI3K function and consequently inhibits downstream signaling through AKT [45], leading to the inhibition of TRP53 degradation. Evidently, knockdown of PTEN releases AKT from its inhibition, initiating the degradation of TRP53 via MDM2 [46]. This suggests that the observed increased Nanog expression after TRP53 and PTEN down-regulation are part of the same mechanism. However, there are 2 indications that TRP53 and PTEN suppress Nanog in SSCs independently from one another. First, knockdown of either gene did not influence the expression of the other, and second, different effects were observed on Ddx4 and Plzf expression levels between the TRP53 knockdown and the PTEN knockdown.
NANOG is also up-regulated upon differentiation of male germ cells (submitted for publication) in a meiotic and post-meiotic expression pattern that is reminiscent of DDX4 expression [28]. DDX4 is an ATP-dependent RNA helicase of the DEAD-box protein family, expression of which is restricted to the germ lineage [27,28,47,48]. Ddx4 expression was unaffected by decreased TRP53 levels, which indicates that elevated Nanog levels were not caused by differentiation of germ cells. Likewise, Ddx4 expression was expressed at significantly lower levels in cells with decreased Pten levels, which excludes the possibility that the increase in Nanog expression reflects the presence of differentiating male germ cells. A decrease in expression levels of Ddx4 upon PTEN down-regulation is in line with the progressive loss of Ddx4 expression in primordial germ cells of Pten null embryos [20]. Post-transcriptional processing of RNA could be one component of the mechanism by which germ cells are insulated from pluripotency.
The degree of up-regulation of Nanog after knockdown of TRP53 or PTEN was still much less than the difference in Nanog expression between ES cells and SSCs cells (Fig. 1). Moreover, forced expression of Nanog or Sox2 in Trp53 −/− SSCs is not sufficient to induce the conversion of these cells to pluripotency [4], which suggests that more factors than Nanog are necessary for a full conversion. The increase in Nanog expression upon TRP53 knockdown is probably a direct effect as TRP53 suppresses the expression of Nanog by binding to its promoter [13]. It is therefore remarkable that expression levels of Nanog and other pluripotency-related genes in Trp53 −/− SSCs are comparable to those of wild-type SSCs [4], which could indicate that these cells are redundant of mechanisms that are involved in suppression of Nanog. This is in agreement with the findings of the current study that PTEN suppresses Nanog independently from TRP53. A surprising finding of the current study is that Trp53 expression levels were approximately equal in ES cells and SSCs, which is contradictory to the direct effect that TRP53 has on the expression of Nanog [13] and its alleged roles in refraining SSCs from pluripotency. This suggests that in mouse ES cells, post-transcriptional processes influence the suppressive activity of TRP53 on Nanog.
Forced expression of c-Myc, Klf4, Oct4, and Sox2 can reprogram somatic cells toward a pluripotent state, a process that can be suppressed by TRP53 [15 –19,29 –32]. Interestingly, Nanog is essential for cells to acquire pluripotency [49], and expression of this gene can be used to distinguish between fully reprogrammed cells and incomplete reprogramming [50], which supports the thought that the observed increase in Nanog expression upon knockdown of TRP53 and PTEN reflects the first step toward reprogramming of SSCs.
We did not observe a change in cell morphology after knockdown of Trp53 and Pten, nor a switch to an ES cell-like transcriptional program, which suggests that the SSCs did not acquire pluripotency. If this would have been otherwise, pluripotency of the cells could have been further assayed by the formation of teratomas and the ability to differentiate into various cell types of 3 germ cell layers.
A surprising finding was the increase in expression of SSC-marker Plzf after TRP53 knockdown, because Plzf is expressed in SSCs rather than in pluripotent cells. PLZF is a transcriptional repressor that regulates the epigenetic state of undifferentiated cells and is required for self-renewal and maintaining the stem cell pool [25,26]. Interestingly, a PLZF-retinoic acid receptor alpha fusion protein has been shown to inhibit TRP53-dependent transcription and leads to degradation of TRP53 [51]. Consequently, increased Plzf expression would further reduce TRP53 protein levels in a feed-forward manner. Little is known about Plzf expression in the germ line, and therefore it is difficult to interpret what the observed increase in expression implies, but intermediate enhanced expression of Plzf in the transitional state from SSCs toward ES-like cells is feasible.
In the current study, no major effects were observed of TRP53 or PTEN knockdown on expression of the reprogramming factors c-Myc, Klf4, and Oct4. Sox2 expression was observed in more samples in which TRP53 was knocked down than in mock control samples. This could indicate that Sox2 participates as a reprogramming factor in the initial events of the transition of SSCs toward ES-like cells. The contribution of the reprogramming factors in the transition from unipotent SSC toward a pluripotent ES-like cell needs to be further investigated. It has recently been reported that colonies of pluripotent cells were more often observed in low-density cultures than in high-density cultures of germ-line stem cells [5]. It is interesting to speculate that an autocrine factor produced by germ-line stem cells insulates these cells from pluripotency, possibly through interaction with TRP53 and/or PTEN.
In summary, the findings of the current study suggest that TRP53 and PTEN have redundant roles in insulating SSCs from pluripotency through suppression of the pluripotency gene Nanog. It would be interesting to test whether ectopic expression of Pten and/or Trp53 in ES cells, over and above their endogenous levels, is sufficient to suppress Nanog expression in ES cells, and thereby trigger their transition to a unipotent state. The results from this study enhance our understanding of the mechanism that insulates pluripotency in SSCs.
Author Disclosure Statement
No competing financial interests exist.