
Abstract
The capacity of the lung to repair itself after injury is well known, but the cell types involved in lung regeneration remain undefined. The aim of this study was to isolate and characterize resident progenitor/stem cells from adult mouse lung. We report the isolation and characterization of resident stem cells that have a Sca1(+)/CD45(-)/CD31(-) phenotype. Their immunophenotype and differentiative potentiality were distinct from that of other previously described lung stem cells. These cells underwent extensive self-renewal in culture and could differentiate into endothelial and lung epithelial (alveolar type I, II, and Clara) cells in vitro. They have exhibited some mesenchymal but no neural differentiation ability. Transfer of these cells into mouse models of lung injury significantly improved survival and minimized lung destruction. These cells may provide useful tools for the study of lung stem cells and the assessment of new therapeutic approaches for lung diseases.
Introduction
E
Materials and Methods
Mice
All animals used in this study were C57BL/6 mice, 6–8 weeks old, maintained in our animal facilities under specific pathogen-free conditions with the approval of the Tohoku University Review Board (Manual for Animal Studies). Mice transgenic for enhanced GFP on a C57BL/6 strain background were established at the Research Institute for Microbial Disease (Osaka University, Japan) [6].
Preparation of mouse embryonic fibroblasts, feeder cells, and feeder-conditioned medium
DR4 mice mouse embryonic fibroblasts (MEFs) were purchased from ATCC or embryos were collected from C57BL/6 pregnant females at day 13 p.c. and washed with PBS; their soft tissues were removed; and embryonic carcasses were minced with a scissor. Then the minced tissue was digested in trypsin at 37°C for 30 min, sequentially passaged through G18, G20, and G23 needles using a syringe, filtered, and cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with pyruvate and glutamine (Invitrogen, Carlsbad, CA). Penicillin/streptomycin, 10% FBS, and 1% nonessential amino acids were added to the medium and cells were allowed to grow to confluence. Since MEFs are primary cells, they have a limited lifespan in culture and were passaged for only 1–5 passages. Confluent MEFs were passaged or treated with 10 µg/mL mitomycin-C (Sigma, St. Louis, MO) for 2 h to arrest cell growth. Mitomycin-C-treated cells were collected following trypsinization and stored in liquid nitrogen until use. Feeders-conditioned medium (FCM) were collected daily from freshly cultured feeder cells and supplemented with equal volumes of fresh medium. Feeder cells were again fed with medium and used for FCM collection for 7–10 days.
Isolation of lung stem cells
Single-cell suspensions were prepared each time from lungs of at least 3 mice as described [7]. In brief, 1 mL dispase I (2 U/mL; Roche Diagnostics, Indianapolis, IN) was injected through the trachea. Subsequently, the lungs were incubated in a 37°C shaking incubator for 45 min in 10 mL of dispase, 1 mL of 0.001% DNAse (Sigma), and 1 mL of 2 µg/mL collagenase/dispase (Roche). The trachea and bronchi were removed, and the lungs were minced and incubated for 10 min. This suspension was filtered on 40-µM filter, centrifuged, and depleted of red blood cells by incubation in RBC lysis buffer (Sigma). Cells were resuspended in PBS at 1 × 106 cells/100 µL and stained prior to analysis using the AutoMACS cell separator system (Miltenyi Biotec, Bergisch Gladbach, Germany) and/or FACSAria cell sorter (Becton Dickinson, Franklin Lakes, NJ).
Flow cytometry and immunofluorescence
To sort the stem cells and conduct analyses on the expression of various surface markers, cells were stained for 15 min with specific antibodies and analyzed by FACS. For FACS analyses, IntraStain (Dako, Glostrup, Denmark) was used for the fixation and permeabilization of cells prior to staining of intracellular markers. For immunofluorescence, cells were plated on coverslips and allowed to adhere overnight. They were then fixed with 4% paraformaldehyde and incubated in 5% goat serum + 5% BSA and 0.1% Triton X-100 to block nonspecific binding and permeabilize the cell membrane, respectively. The cells were then stained with primary and secondary antibodies according to standard procedures. For immunofluorescence staining of lung tissue, lungs were fixed in 4% paraformaldehyde, cryoprotected in 10% and then 20% sucrose, and embedded in O.C.T. (Sakura Finetek). Three-micron cryosections were blocked, permeabilized, and stained with primary and secondary antibodies. Slides were mounted with VECTAshield containing DAPI (Vector Laboratories, Burlingame, CA) and viewed on a fluorescent microscope (Leica, Voorburg, The Netherlands). The anti-mouse antibodies used for flow cytometry and immunofluorescence included the following: Sca1-APC, CD45-PE, CD31-PE, CD34-FITC, CD44-PE and CD106-FITC, surfactant protein-C (SP-C), and CD31 (BD Pharmingen, San Diego, CA); CD45 and CD34 (Chemicon, Temecula, CA); Sca1 (R&D Systems, Minneapolis, MN); goat anti-mouse aquaporin-5, vimentin, alpha smooth muscle actin (αSMA), and CC10 (Santa Cruz, Biotechnology, Santa Cruz, CA), and GFP (Abcam, Cambridge, MA). Goat anti-rabbit and anti-rat and donkey anti-goat and anti-mouse Alexa Flour and Q-dot (Invitrogen) were used as secondary antibodies for double and triple immunofluorescence staining. All staining procedures were run with negative controls and the appropriate isotype controls (eBioscience and Biolegend).
Limiting dilution analysis
Limiting dilution analysis (LDA) was performed as descried [8] with modifications. The exact number of cells with colony-forming ability within a putative stem cell population cannot be determined by culturing large number of cells together and counting number of colonies later because stem cells replicate and give rise to progenitors or daughter stem cells, which, in turn, can give rise to multiple colonies. Various dilutions of cell suspensions were plated into well plates for an appropriate culture period. Even when wells contained an average of 10 cells, all wells contained colonies indicating a very high colony-forming ability. So we diluted our cells to 1 cell per 100 µL and dispensed 100 µL/well of three 96-well plates. Then, we examined all wells under microscope for the number of cells in each well. Empty wells or wells that contained >1 cell were ignored while wells that contained single cells were marked and examined daily to observe their colony-forming ability. This was repeated at least 3 times for cells from different batches and passage number. Some colonies that originated from single cells were dissociated and re-cultured in another set of 96-well plates to examine for the cell ability to produce secondary and tertiary colonies.
Semiquantitative RT-PCR
Total RNA was extracted from stem cells and differentiated cells with an RNAeasy kit (Qiagen, Tokyo, Japan). The RNA concentrations were determined and the samples were diluted to 50 µg/mL. Reverse transcription and PCRs were conducted in one step using a one-step RT-PCR kit (Qiagen). The optimal cycle numbers and annealing temperatures were determined for each set of primers before all of the samples were compared. b-Actin was used as an internal control.
Cell culture and induction of multipotent differentiation
Cells were plated in high-glucose DMEM supplemented with pyruvate and glutamine (Invitrogen). Penicillin/streptomycin, 10% FBS, and nonessential amino acids were added to the growth medium. Feeder cells do not proliferate and we found that when they are passaged for a second time, almost all of them are unable to attach to culture plates. On passaging the growing colonies on feeder cells, we trypsinized and collected all cells and passaged a portion of them to the new culture plate. Any older feeder cells will not attach to the new plate and will be washed away with the first medium change. Near confluent cells were trypsinized prior to passaging or in preparation for FACS analysis or RNA collection. For differentiation studies, cells were seeded on various combinations of thin/thick Matrigel (BD Bioscience, San Jose, CA), mixed with the gel before it hardened or seeded on its top after hardening in SFM/DMEM/FCM supplemented with 20–40 ng/mL human recombinant HGF (gift from Prof. Nakamura, Osaka), 40–100 ng/mL EGF (Sigma), or 20–100 ng/mL bFGF (Biosource). The medium was replaced every 2 or 3 days. Differentiation into the various mesenchymal cell types was induced using the “mesenchymal stem cell functional identification kit” according to the manufacturer’s protocols (R&D Systems). For adipogenic differentiation, MLSCs were cultured in aMEM basal medium until 100% confluency is reached. Then medium was replaced with adipogenic differentiation medium. Lipid droplets and signet ring cells were observed after 10 days. Then, cells were either fixed and stained with Oil Red or collected for RNA separation. For osteogenic differentiation, MLSCs were cultured in aMEM basal medium until 50%–70% confluent. Then medium was replaced with osteogenic differentiation. After 21 days (when cells started to detach), cells were either stained with Alizarin Red S or collected for RNA. For chondrogenic differentiation, 250,000 MLSC were suspended in 0.5 mL of chondrogenic differentiation medium and centrifuged. Medium was not removed after centrifugation and tubes were incubated upright at 37°C and 5% CO2. After 21 days, cell pellets were fixed and cut as frozen sections that were subjected to Alizarin Blue staining or RNA was isolated from them for RT-PCR examination. Musculogenic differentiation was induced by incubating near confluent cells with medium containing 5 mM 5-Azacytidine (Sigma) for 24 h. Cells were then washed and maintained in regular medium. The presence of desmin (a pan-muscle marker) was detected 10 days later by RT-PCR and immunofluorescent staining.
Immunofluorescence staining of MLSC differentiated on Matrigel
Differentiated cells and structures were released from the Matrigel by mixing the gel with ice-cold saline that caused the gel to liquify without enzymatic digestion so that the differentiated structures would remain intact as much as possible. Cells were then spun into glass slides and stored frozen or stained immediately. Cells were labeled for different combinations of CD31, SP-C, aquaporin-5, CC10, and/or Sca1. In other set of experiments, differentiated cells on Matrigel were dissociated into single-cell suspensions using “Cell Dissociation Solution” (BD Bioscience) and used for RNA isolation, flow cytometry, or immunofluorescence.
In vivo study
Animal treatment with elastase, lung histopathology, immunofluorescence and quantitation of emphysema, and GFP(+) engraftment were carried out as previously described with modifications [7]. In brief, the mice were anesthetized and the trachea exposed by blunt dissection. Elastase, cells suspended in 10% serum-enriched DMEM or 10% serum-enriched DMEM alone, were delivered via a 26-gauge needle inserted directly into the tracheae of mice from the different groups described in legends of Figure 5. After the animals were killed, the trachea and lungs were removed together and inflated with 4% paraformaldehyde at a pressure of 20 cm H2O. The tissue was fixed and 3-mm thick sections were stained with hematoxylin and eosin. Air space enlargement was quantified stereometrically using the mean linear intercept (L m) method in 20 randomly selected fields of tissue sections. L m was determined by counting alveolar wall intersections with an array of test grid lines drawn horizontally on a printout of the digital image of each section and calculated according to the equation: L m = 2L T/I w, where I w is the number of times the test line intersected with an alveolar wall and L T is the length of the test line.
Statistics
Data are expressed as the mean ± SEM. Statistical analysis was performed on a Macintosh computer system with JMP 7 statistical analysis software (SAS Institute, Cary, NC). Normally distributed data were assessed for significance by ANOVA, followed by Student’s t-test for multiple comparisons. Differences in survival among the treatment groups were assessed using the Kaplan-Meier method with log-rank and Wilcoxon tests. A difference was considered statistically significant at P < 0.05.
Results
Isolation of potential stem cell population from mouse lung
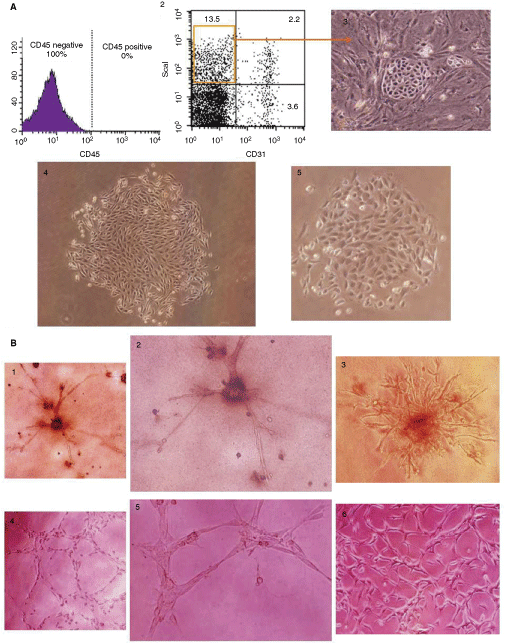
Based on the previous works [2,3], we speculated that an adult lung stem cell population resided in the Sca1(+)/CD45(−)/CD31(−) cell population. To investigate this hypothesis, single-cell suspensions were prepared from the lungs of C57BL/6 mice. Due to the high number of CD45(+) cells in the suspensions, mouse anti-CD45 beads were employed to deplete CD45(+) cells from the suspensions using the AutoMACS system. Subsequently, Sca1(+)/CD45(−)/CD31(−) cells were sorted out of the CD45(−) fraction using FACSAria (Fig. 1A-1 and 1A-2). Sorted cells were cultured on feeder cells or in chambers above feeder cells (to avoid cell–cell contact with the feeder cells) using serum-enriched DMEM. Additionally, some cells were cultured on uncoated regular tissue culture plates using DMEM or FCM.
(
Growing colonies could be detected in all the examined culture conditions. However, culture on feeder cells was the earliest assessed condition to support the growth of colonies. They were first observed after 6–7 days (Fig. 1A-3) and became confluent within 10–12 days. In addition to growing colonies, several morphologically heterogeneous cells could also be seen growing on the feeder cells until passage 5 or 6. From then on, the short spindle-shaped colony-forming cells were predominantly observed and the cultures could be split at higher ratios.
Colonies resulting from cells plated in chambers above feeder cells or on regular plates without feeder cells, with or without FCM, took longer time to show up (10–12 days) and to reach confluence (18–24 days) and were fewer in number compared to colonies growing on feeders cells. These colonies were accompanied by much more morphologically heterogeneous cells than those observed with the colonies growing on feeder cells. However, repeated passages eventually caused disappearance of the heterogeneously growing cells and predominance of the short spindle-shaped colony-forming cells. These colonies were exactly similar to those growing on feeder cells although they took much longer time. These observations suggest that the presence of feeder cells is advantageous but not essential for lung progenitor cell survival and growth in primary culture. They seem to support and speed up the proliferation of the very rare progenitor cells within the abundant nonprogenitor cell populations resulting in earlier takeover. To further confirm this, progenitor cell colonies that were grown on feeder cells till passage 6 and later were passaged on top of feeder cells, into chambers above feeder cells, or into regular tissue culture plates with FCM or DMEM. The cells grown under all assessed culture conditions formed colonies, but the rates of growth varied. Feeder cells enabled the fastest proliferation, followed by the chambers above feeders, then tissue culture plates with FCM. Cells that were passaged into regular culture plates with serum-enriched DMEM were the slowest to form colonies (Fig. 1A-4). Culture with serum-free medium (SFM) arrested colony growth and re-use of FCM or serum-enriched DMEM re-established growth, indicating the need for unidentified growth factors provided by the feeder cells or serum to induce and maintain the clonal growth.
The capacity of our primary Sca1(+)/CD45(−)/CD31(−) cells to form colonies was 0.02% on feeder cells and 0.01% or less on other culture conditions. Seeding at a density lower than 0.5 × 104 cells/mL usually failed to produce colonies on feeder cells, while densities of 1 × 104 cells/mL or higher were needed for chambers above feeder cells or on regular plates without feeder cells, with or without FCM. We can usually retrieve ∼5 × 106 cells from 1 lung and 74 or less primary colonies could be found from one lung.
MLSCs undergo extensive self-renewal
A key feature of stem cells is their ability to maintain stem cell properties after division, a feature known as self-renewal. When single-cell suspensions of the putative stem cell colonies were cultured at clonal density (1,000 cells/well), all wells gave rise to colonies within 6 days. To exclude the possibility of overestimation due to cell aggregation or formation of secondary colonies, LDA was performed by culturing single cells in individual wells of 96-well plates. We found that 27% ± 3% of the wells contained colonies. Importantly, seeding of single cells that were generated from secondary clones led to the generation of tertiary clones, further confirming their self-renewal ability. Daughter colonies were always morphologically (Fig. 1A-5) and phenotypically (Figs 2 –4) identical to primary cell colonies. Of the 6 established MLSC lines, 2 were maintained in culture for >12 months and passaged >50 times without losing their colony-forming ability. From these cultures, individual clones were derived from single cells by limiting dilution. One clone from each line was further characterized.

Confirming the identity of the differentiated cells in the Matrigel. The differentiated colonies and cells described in Figure 1B were collected from Matrigel after 10–15 days and dissociated into single-cell suspension. Roughly half of the cells were used for RNA extraction whereas the other half was used for immunofluorescent staining and FACS analysis. Immunofluorescent staining was performed on Cytospins or cells that were cultured in chamber slides for 2–3 h after collection. RT-PCR of RNA from undifferentiated multipotent lung stem cells (MLSCs) before culturing them on Matrigel indicated that the MLSCs have weak baseline expression of several alveolar epithelial, Clara, and endothelial cell markers and high expression of mesenchymal cell markers (“before differentiation” bands in
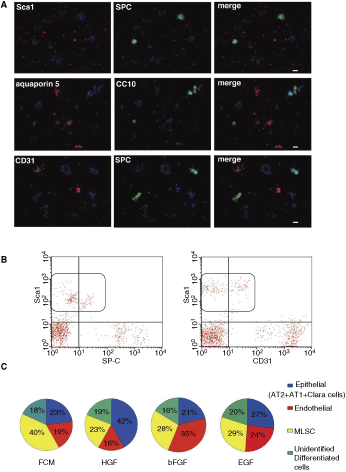
Characterization and quantification of cells differentiated in Matrigel from whole multipotent lung stem cells (MLSCs). Differentiated colonies and structures described in Figures 1B were partly dissociated from the Matrigel after 10–15 days and immunofluorescently labeled for different combinations of CD31, SP-C, aquaporin-5, CC10, and/or Sca1. The appropriate isotype controls were used concomitantly as shown in Figure 2B-1, B-2, and B-3. (
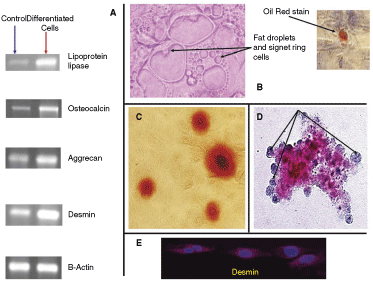
multipotent lung stem cells (MLSCs) differentiate into cells of mesenchymal origin. MLSC at passages 10, 21, and 30 (result of cells from passage 21 are shown) were induced to differentiate into adipocytes, osteoblasts, chondroblasts, and muscle cells by incubation with defined factors. (
Clones were cryopreserved at passages 3, 14, 20, 30, and 39. When these cells were re-cultured, no differences in growth kinetics or morphological or functional characteristics were observed between stem cell clones derived from cryopreserved and never-frozen cells. Our identification of a lung cell population with a phenotype suggestive of stem cells prompted us to determine whether these cells showed functional properties associated with lung stem cells.
MLSCs generate endothelial and alveolar cells in vitro
First, MLSCs were tested for their capacity to differentiate into any of the lung cell lineages in vitro. Cells from 2 different clones at passages 9, 18, 27, and 35 were cultured on Matrigel. Various combinations of methods for the induction of differentiation were evaluated (see Materials and Methods). We found that culturing MLSC embedded inside Matrigel induced the cells to form branches and tube-like extensions that resembled endothelial capillaries (Fig. 1B-1 to 1B-3) and that supplementing the Matrigel and medium with 20 ng/mL basic fibroblast growth factor (bFGF) potentiated this differentiation. MLSC that were attached to the plate bottom below the Matrigel formed complex structures that were morphologically similar to a 2-dimensional section through lung tissue (Fig. 1B-4 to 1B-6) and supplementing with 20 ng/mL hepatocyte growth factor (HGF) potentiated this differentiation. Culturing in Matrigel supplemented with 40 ng/mL epidermal growth factor (EGF) potentiated the production of both morphologies.
To further characterize the identities of these differentiated cells, they were dissociated from Matrigel after 10–15 days into single-cell suspensions and further evaluated by RT-PCR, immunofluorescence staining, and flow cytometry. As seen in Figure 2B and in the “before differentiation” bands of all RT-PCR experiments in Figure 2A, before culturing them on Matrigel, undifferentiated MLSC cells weakly expressed almost all of the evaluated epithelial and endothelial markers. They also strongly expressed the mesenchymal markers vimentin and αSMA. Cells differentiated on Matrigel showed a marked induction of their RNA expression for the various lung epithelial and endothelial cell lineage markers and a marked reduction in the RNA levels of the mesenchymal markers vimentin and αSMA (Fig. 2A). Immunofluorescence analyses confirmed cellular differentiation by showing that many of the MLSCs had lost their concomitant weak expression of multiple lineage markers and strongly expressed markers of only a single lineage (ie, AT-I, AT-II, Clara, or endothelial cells) (Fig. 2C).
Then, we wanted to examine how perfect these differentiated structures resembled the normal alveoli. In other words, we wanted to examine if these differentiated structures appropriately consisted of an anatomically relevant organization of alveolar types I and II cells, in apposition to endothelial-like cells or not. Double or triple immunofluorescence stainings were conducted on several differentiated structures (like those shown in Fig. 1B-1 to 1B-6) after partial dissociation from the Matrigel to assess the relative contribution of each marker within the structure. We found that all cells within most structures strongly expressed only one single lineage marker, that is they were purely SP-C(+), aquaporin-5(+), or CD31(+) (Fig. 3A) and only very few structures contained combinations of 2 or more markers. This means that under the described differentiation conditions, each MLSC differentiated into pure endothelial or pure alveolar cells, not into a perfect alveolar unit that contained alveolar and endothelial cells.
FACS was regularly used to quantify the types and percentages of differentiated and undifferentiated cells that dissociated from the Matrigel. Cells that were still expressing Sca1 were considered undifferentiated MLSCs, whereas cells that were Sca1(−) and SP-C(+), aquaporin-5(+), CC10(+), or CD31(+) were considered AT-I, AT-II, Clara, or endothelial cells, respectively. Cells that were Sca1(−) and lineage marker(−) were considered unidentified differentiated cells. Although different batches of MLSCs with different passage number varied in their differentiation efficiencies, MLSCs differentiated on Matrigel plus 20 ng/mL HGF consistently produced 30%–50% differentiated epithelial cells (ie, AT-II, AT-I, and Clara cells), 10%–20% endothelial cells, and 10%–30% MLSCs; the rest were unidentified differentiated cells. MLSCs differentiated on Matrigel plus 20 ng/mL bFGF consistently produced 25%–40% differentiated endothelial cells, 15%–25% epithelial cells (ie, AT-II, AT-I, and Clara cells), and 20%–30% MLSC; the rest were unidentified differentiated cells. Matrigel plus 40 ng/mL EGF produced 20%–25% differentiated endothelial cells, 25%–30% epithelial cells (ie, AT-II, AT-I, and Clara cells), and 30%–35% MLSC; the rest were unidentified differentiated cells (n = 6, Fig. 3B and 3C).
To prove that a single stem cell could differentiate into both lineages, other similar sets of differentiation studies were conducted on clones that were originated from a single cell by LDA. They produced exactly similar results (n = 3, data not shown). These data demonstrate that MLSC can generate both endothelial and alveolar cells in vitro.
MLSCs show mesenchymal but not neural differentiation potential in vitro
We examined the ability of our cells to differentiate into cells of mesenchymal origin. Using a commercial kit, adipogenic, chondrogenic, osteogenic, and musculogenic differentiation was induced. After incubation of 100% confluent cells in adipogenic differentiation medium for 10 days, fat droplets and signet ring cells were observed and stained positive with Oil Red (Fig. 4B). Marked induction of lipoprotein lipase gene expression was detected by RT-PCR (Fig. 4A). When 50% confluent cells were incubated for 21 days in osteogenic differentiation medium, several cells started to detach and many stained positive with Alizarin Red S (Fig. 4C). Marked induction of osteocalcin gene expression was detected by RT-PCR (Fig. 4A). Chondrogenic differentiation was induced by culturing 250,000 pelleted cells in a 15-mL conical tube with chondrogenic differentiation medium for 21 days. The cell pellets were collected and their frozen sections were subjected to Alizarin Blue staining. Portions of the pellet sections showed positive staining for Alizarin Blue (Fig. 4D). Marked induction of aggrecan gene expression was detected by RT-PCR (Fig. 4A). Musculogenic differentiation was induced by incubating the cells with medium containing 5 µM of 5-Azacytidine for 24 h. Cells were then washed and maintained in regular medium. The presence of desmin (a pan-muscle marker) was detected 10 days later by RT-PCR (Fig. 4A) and immunofluorescent staining (Fig. 4E).
Although culturing MLSCs on uncoated plates with SFM containing 40 ng/mL bFGF and 20 ng/mL EGF induced them to form sphere-like structures that were morphologically similar to neurospheres, there was no significant induction of any of the tested neural cell markers, including nestin, b-tubulin, NG2, CNP, and GFAP (data not shown). The pattern of differentiation of MLSCs into the epithelial and mesenchymal lineages remained fairly stable up to passage 35. However, although cells with higher passage numbers continued to demonstrate the colony-forming ability, their differentiation studies started to take longer and the resulting differentiated/undifferentiated cells were of lower ratios than the more early passages.
Isolation of lineage-negative MLSCs
The previously described BASCs [2] were lineage-positive because they expressed SP-C and CC10, which suggests that they are more likely progenitors rather than stem cells. BASCs [2] were isolated based on being Sca1(+), CD34(+), CD45(−), and CD31(−). They had been passaged only 9 times and the expression status of Sca1 and CD34 after several passages in culture was not discussed [2]. Our MLSCs as well were lineage-positive because they weakly expressed several epithelial, endothelial, and mesenchymal lineage markers, including SP-C and CC10 (Fig. 2). We hypothesized that MLSCs may be pushed up the “stemness axis” by changing the medium type and/or serum concentration. We cultured MLSCs in several different conditions (Table 1) and assessed lineage expression by RT-PCR. We found that culturing cells with DMEM + 0.5% FCS was the best condition that caused all lineage marker expression to disappear.
E
Abbreviations: GF, growth factor; FGF, basic fibroblast growth factor; EGF, epidermal growth factor.
These lineage-negative MLSCs retained all their stem cell characteristics. They continued to self-renew, although at a slower rate, and they showed improved colony formation in LDA (70% compared with 27% in lineage-positive MLSC; for BASCs [2], only 1 cell of 81 cells formed a colony). When lineage-negative MLSCs were exposed to sets of differentiation studies similar to these conducted on lineage-positive MLSC, they showed similar or even faster multipotent differentiation ability. Raising the serum concentration back into 5%–10%, however, induced faster growth rates and re-expression of the various lineage markers. Lineage-negative MLSCs cultured in DMEM + 0.5% FCS were nearly always 100% Sca1(+) and CD34(+). Lineage-positive MLSCs grown in DMEM + 10% FCS had variable levels of Sca1 and CD34 expression, depending on the batch and passage number. Sca1 expression ranged from 50% to 100% and CD34 expression was from 60% to 100%. Sca1(+)/CD34(+) cells were 40%–80%.
Because our MLSCs expressed the mesenchymal markers vimentin and αSMA and showed mesenchymal differentiation characteristics, we examined their similarity to the mesenchymal progenitor cells isolated from the adult mouse lung side population [3] that were Sca1(+), CD44(+), CD106(+), CD45(−), and CD31(−). 20%–70% of our MLSCs expressed CD44, 5%–50% expressed CD106, and 5%–40% were double-positive, depending on the batch and passage number. Sorting of the Sca1(+), CD44(+), and CD106(+) cells out of the MLSC population did not improve the MLSC mesenchymal differentiation characteristics and the percentage of CD44(+) and CD106(+) cells fell rapidly after several passages (data not shown). Changing the medium type and/or serum concentration failed to bring CD44 and CD106 expression to 100% or improve the MLSC mesenchymal differentiation characteristics. We conclude that MLSCs are a different stem cell population than lung MSCs [3].
MLSCs show minimal engraftment in the uninjured lung
To determine whether lung stem cells are also multipotent in vivo, 0.5 × 106 cells from a passage 9 MLSCs isolated from GFP transgenic mice were administered intratracheally to 15 wild-type mice. We used cells grown from a single colony that was derived from a single stem cell by LDA. Three mice of each time point were sacrificed on days 1, 4, 10, 20, and 30, and their lungs were examined for GFP(+) cells. On day 1, many MLSCs were seen inside the alveoli and conducting airways. These cells were rounded, undifferentiated, and continued to express Sca1 (Fig. 5A). From day 4 onward, very few GFP(+) cells could be detected in the epithelial lining of the airways and alveoli. We also observed gradual clearance of GFP(+) cells from the airway and alveolar lumens. Double staining with markers of lung epithelial cell lineages showed very few alveolar type 2 cells (Fig. 5B) and Clara cells (Fig. 5C) that were GFP(+). Mice that were given MEFs as controls never showed any engraftment.
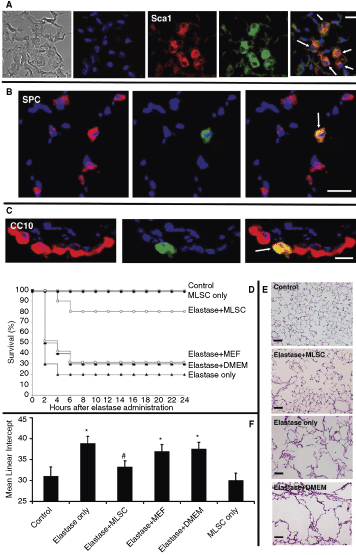
Intratracheal administration of multipotent lung stem cells (MLSCs) to uninjured and injured lungs. (
MLSCs improve survival and exert a protective effect against elastase-induced lung injury and emphysema
Then we wanted to examine the ability of MLSCs to engraft in an injured lung and whether such engraftment would potentiate survival and healing of lung injury. First, we examined the effect of administering MLSCs intratracheally on the survival of mice receiving elastase. Mice were given a dose of intratracheal elastase that caused 80%–100% lethality within 4 h. Treatment of mice with MLSCs 30–60 min after elastase administration significantly improved survival compared to mice that received elastase only or elastase followed by DMEM or MEFs (used as controls for the MLSC vehicle and cell effect, respectively) (Fig. 5D). Surviving mice were sacrificed at 24 h. No MLSC engraftment was observed indicating that their survival improving effect must have been through an immunomodulatory mechanism that inhibited the elastase-induced acute lung injury and lethality.
Second, we examined the effect of MLSCs on the development of emphysema in mice receiving a sublethal dose of elastase that produced massive pulmonary emphysema in 100% of mice within 3 weeks. Mice were given MLSCs 30–60 min after intratracheal administration of elastase and the lungs were histologically examined 3 weeks later. Mice treated with MLSCs had significantly less elastase-induced enlargement of air spaces and alveolar wall destruction compared with the elastase only or elastase + DMEM-treated mice (Fig. 5E). In the elastase + MLSCs group, we observed only a 4% increase in the widely used emphysema assessment quantitative parameter, the L m. However, 25% and 21% increases were found in the elastase only and elastase + DMEM groups, respectively (Fig. 5F). These results indicate that cell therapy with MLSCs has a protective effect against the development of emphysema in elastase-injured lungs. Only minimal MLSC engraftment was observed in mice sacrificed at 3 weeks after treatment. Quantitative assessment of GFP(+) cells co-localization with the various lineage markers showed that MLSCs co-localized only with alveolar type 2 cells and that the GFP(+)/SP-C(+) cells were only 1% of the total SP-C(+) cells. This again indicates that the MLSC protective effect on emphysema development must have been through an immunomodulatory mechanism that inhibited the elastase-induced acute lung injury and destruction. This very low engraftment in uninjured and injured lungs is in line with what have been described recently for embryonic and MSCs [9]. Taken together, these data indicate that MLSCs can give rise to endothelial and lung epithelial cells in vitro and that cell therapy with MLSCs can improve survival and minimize lung damage after injury.
Discussion
In this study, we describe a reproducible method for isolating, purifying, and maintaining a population of multipotent stem cells from the mouse lung. These stem cells undergo extensive self-renewal in culture; can generate endothelial, Clara, AT-I, AT-II cells; and can differentiate into cells of mesenchymal origin in vitro. We also showed for the first time that a population of mouse resident lung cells could ameliorate various pathological outcomes subsequent to elastase-induced lung injury.
Increasing evidence suggests that the differentiation potential of adult stem cells may extend to lineages beyond those usually associated with their germ layer of origin [10]. Some tissue stem cells show great plasticity beyond the lineages for which they were first characterized and their multipotentiality and even pluripotentiality have been described [11]. The ability of MLSCs to differentiate into cells of endodermal and mesodermal origins is not unique because tissue stem cells that express both epithelial and mesenchymal markers have been described [12 –14]. For example, kidney papillary progenitor cells have been shown to co-express both epithelial and mesenchymal proteins and give rise to myofibroblasts and cells expressing neuronal markers [12]. Also, murine liver resident stem cells have been differentiated into hepatocytes, cholangiocytes, osteoblasts/osteocytes, chondrocytes, astrocytes, and neural cells [13]. In addition, resident multipotent progenitor cells in adult human glomeruli have been shown to express vimentin and cytokeratin [14].
The lack of specific marker(s) that are uniquely expressed by lung stem cells has long hampered their in vivo localization and tracing as well as their prospective isolation and characterization. Although we are still unable, at this stage, to identify the exact anatomical location of MLSCs, our reproducible methods for isolating and purifying the lineage-negative MLSCs should allow for the detection and verification of such marker(s) in the near future. It is worth noting that the SP-C and CC10 double-positive cells that are usually thought of as BASCs [2] and that its niche is described to be the bronchioalveolar junction were later detected in ectopic locations that include the alveolar space and sub-bronchiolar regions [15,16]. This finding highlights the pressing need to identify specific and unique markers for lung stem cells. Alvarez et al. isolated progenitor cells from rat lung microvascular endothelium that were capable of proliferation and colony formation in culture and exhibited tube formation on Matrigel within 24 h. These cells were CD31(+) and reached confluence easily on tissue culture plates in DMEM [17]. To the contrary, our cells were isolated from murine lung cells and were primarily CD31(−), Sca1(+), expressed several specific alveolar cell markers, and needed at least 10 day to reach confluence on feeder cells and 5–10 days to exhibit the tube formation on Matrigel.
The continuous high level of vimentin and αSMA expression in MLSCs and partial expression of other MSC markers, such as CD44 and CD106, prompted us to explore their potential as endogenous mesenchymal progenitor cells and to compare them with the cells described by Summer et al. [3]. We were not able to induce our MLSCs to assume complete MSC characteristics in vitro; however, our ability to promote MLSCs to partially differentiate into several mesenchymal phenotypes indicates that MLSCs may have an important role in maintaining the lung parenchyma. MSC have been used widely in both autologous and allogeneic cell therapy studies. Despite initial interest in their multipotent properties, organ engraftment did not appear to play a major role. The beneficial effects of MSCs in many organs derived from their capacity to secrete paracrine soluble factors that modulate immune responses as well as alter the responses of endothelium or epithelium to injury through the release of growth factors [18]. Gupta et al. showed increased survival of mice following intratracheal administration of MSCs 4 hours after intrapulmonary administration of a lethal dose of Escherichia coli endotoxin [19]. The beneficial effect was independent to MSC engraftment and the ability of the MSCs to clear the endotoxin. Rather, the beneficial effect was associated with a down-regulation of the proinflammatory and up-regulation of the anti-inflammatory response within the bronchioalveolar lavage and plasma [19]. Acute lung injury and inflammation develops immediately after elastase administration to the murine lung [20]. Ishii et al. showed significant increase in hemoglobin content, albumin concentration, number of neutrophils, number of macrophages, and neutrophil elastase activity in BAL fluids 1 day after elastase administration well before any structural damage could be detected [20]. In this study, we have shown that treatment of mice with MLSCs 30–60 min after administration of a lethal dose of elastase significantly improved survival and that treatment of mice with MLSCs 30–60 min after administration of an emphysema-producing dose of elastase significantly protected the lung from development of emphysema even though MLSC engraftment was very rare. Taken together, we speculate that the mechanism by which MLSC have improved survival and protected the lung from development of emphysema must have been through an anti-inflammatory effect. However, we acknowledge that the exact identification of pathways involved requires further investigation. MLSC in vitro differentiation potential suggests that they may be having a differentiation/regeneration role in vivo. However, the acute lung injury model we used and the early timing of cell administration are not suitable for exploration of this role. Future studies using more chronic injury and later cell administration models may elucidate a lung regenerative role in which MLSCs differentiate to replace and heal damaged lung.
Because any cell therapy with multipotent stem cells will always have a risk of tumor formation, we meticulously observed the lungs of mice that received MLSCs for evidence of tumor formation. After several initial pilot studies, during which we adjusted the relation between number of administered cells and volume of vehicle to avoid cell clumps that may be seen when too large number of cells are loaded in too small volume of vehicle and will be mistaken for tumor growth, we did not detect any evidence of tumor formation in the lung and were unable to detect GFP(+) cells in other organs, for example liver, spleen, and kidney (data not shown). Given that injection of undifferentiated mouse embryonic stem cells into the mouse heart produces a large tumor in 4 weeks [21], we posit that our maximum period of observation (30 days) may not have been sufficient to completely exclude the formation of a tumor inside the lung. We acknowledge that this point requires further study.
Although the lung is an easily accessible organ through the airways, trials for the treatment of intractable lung diseases using stem cell-based therapies are scarce compared with those of other internal organs, such as the heart and brain [22,23]. To our knowledge, this is the first study to evaluate endogenous lung stem cells as a cell-based therapy. Accurate assessment of the degree of survival of MLSC in vivo, the cells’ exact role in the healing process and their cooperation and potentiation function in relation to bone marrow-derived and endogenous lung stem cells are areas that still need to be explored.
In summary, we established multipotent lung resident stem cells from murine lungs from a Sca1(+)/CD45(−)/CD31(−) population. We show that these cells undergo extensive self-renewal in culture and can differentiate into endothelial and lung epithelial cells in vitro. Transfer of these cells into mouse models of lung injury significantly improved survival and minimized lung destruction. These findings have important implications for understanding normal lung homeostasis and response to injury and support the prospect of cell-based lung therapies.
Footnotes
Acknowledgments
This work was supported by a grant from the Japan Society for the Promotion of Science No. 19390222 to H.K. and No. 20890015 to A.E.H. We thank Prof. Masaru Okabe (Genome Information Research Center, Osaka University, Japan) for providing GFP transgenic mice (C57BL/6 TgN(act-GFP)OsbC14-Y01-FM131).
Author Disclosure Statement
No competing financial interests exist.