
Abstract
We have recently identified 2 distinct CD271brightMSCA-1dimCD56+ and CD271brightMSCA-1brightCD56− MSC subsets in primary femur-derived bone marrow (BM), which differ in their expression pattern and morphology as well as in their clonogenic and differentiation capacity. Here, we show that MSCA-1 is identical to tissue non-specific alkaline phosphatase (TNAP), an ectoenzyme known to be expressed at high levels in liver, bone, and kidney as well as in embryonic stem (ES) cells. SDS-PAGE of WERI-RB-1 cell lysate and supernatant from phosphatidylinositol-specific phospholipase C (PI-PLC)-treated WERI-RB-1 cells resulted in the appearance of a prominent 68-kDa band. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI TOF MS) sequence analysis revealed TNAP-specific peptides. Screening of the MSCA-1-specific antibody W8B2 on HEK-293 cells transfected with the full-length coding sequence of TNAP showed specific reactivity with transfected but not with parent cell line. In addition, TNAP-specific mRNA expression was selectively detected in the transfectant line. In agreement with these findings, enzymatic activity of TNAP was exclusively detected in sorted MSCA-1+ BM cells but not in the MSCA-1− negative fraction. Surface marker analysis revealed coexpression of the embryonic marker SSEA-3 but not SSEA-4, TRA-1-60, and TRA-1-81. In endometrium, TNAP is expressed at intermediate levels on CD146+ cells and at high levels in the luminal space of glandular epithelia. Our results demonstrate that TNAP is a selective marker for the prospective isolation of BM-derived MSC and MSC-like cells in endometrium.
Introduction
M
In common protocols, unfractionated bone marrow-derived cells are used as the starting population for the culture of MSC. This isolation method is based on the adherence of fibroblast-like cells to the plastic surface and the removal of nonadherent hematopoietic cells [1,2,4,8]. The resulting cells are poorly defined and give rise not only to heterogeneous MSC populations but also to osteoblasts and/or osteopro-genitor cells, fat cells, reticular cells, macrophages, and endothelial cells [9,10]. To define the starting population more precisely, surface markers such as SSEA-4, the ganglioside GD2, CD140b, CD146, CD200, CD271, the αv/β5 integrin complex, as well as several antibody-defined molecules were used for the prospective isolation of MSC [11 –17]. The latest markers include MSCA-1 and CD56, which have been proved to be suitable surface molecules to purify and characterize MSC subsets from primary bone marrow [18].
Tissue non-specific alkaline phosphatase (TNAP) belongs to a large family of dimeric enzymes common to all organisms. They catalyze the hydrolysis of phosphomonoesters with release of inorganic phosphate (Pi) [19,20]. TNAP is an ectoenzyme anchored to the membrane via a covalent linkage to phosphatidylinositol (PI) that can be cleaved by bacterial PI phospholipase C (PI-PLC) [21,22]. Four isoforms of this enzyme family are described including intestinal, placental, placenta-like, and tissue non-specific phosphatases [19,20]. TNAP is present in bone, liver, kidney, and endometrium [23 –26] and is expressed at high levels on the surface of embryonal carcinoma (EC) and embryonic stem (ES) cells [27]. During differentiation of ES cells, the expression level of TNAP decreases. In the present work, we describe that MSCA-1 is identical to TNAP. In addition, we show differential expression of this ectoenzyme in CD56+ and CD56− bone marrow MSC subsets in the hip joint, as well as in glandular epithelia and MSC-like cells of the endometrium.
Materials and Methods
Primary cells and tissues
Bone marrow cells
Bone marrow from femur, acetabulum, trochanter, and trabeculum was harvested at the Hospital for Workers Compensation from the femoral shafts of patients undergoing total hip replacement. Cells were collected in 5,000 U heparin (Sigma-Aldrich, Taufkirchen, Germany) after informed consent and approval of the Ethics Committee of the University of Tübingen. Bone marrow mononuclear cells were isolated by Ficoll Histopac density gradient fractionation and remaining erythrocytes were lysed in ammonium chloride solution.
Endometrium
Endometrial tissues (n = 11) were collected from women (aged 42.7 ± 4.6 years [±SD], range 36–52 years) undergoing hysterectomy, who had not received hormonal treatment 3 months prior to surgery. Ethics approval was obtained from the Southern Health Human Research and Ethics Committee C and informed written consent was obtained from each patient. Menstrual phase was assessed by histological examination according to well-established criteria; proliferative (n = 4), secretory (n = 5), weakly proliferative (n = 1), and inactive stage (n = 2). Full thickness endometrium with 5 mm myometrium was collected in medium containing HEPES-buffered Dulbecco's modified Eagle's medium/Hams F-12 (DMEM/F12; Invitrogen, Carlsbad, CA), 5% newborn calf serum (CSL, Parkville, Australia), and 1% antibiotic antimycotic solution (final concentrations: 100 mg/mL penicillin G sodium, 100 mg/mL streptomycin sulfate, 0.25 mg/mL amphotericin B; Invitrogen) and processed within 2–24 h, or frozen in optimum cutting temperature (OCT) Tissue Tek® (Sakura Finetek Co., Tokyo, Japan) on dry ice and stored at −80°C until required.
Isolation of W8B2+ cells by MACS and FACS
In selected experiments, the cells were pre-enriched by magnetic-activated cell sorting (MACS) using the W8B2-APC microbead kit (Miltenyi Biotec, Bergisch Gladbach, Germany), according to the protocol provided by the manufacturer. In brief, at least 108 Ficoll-isolated BM cells were incubated with 100 μL of W8B2-APC antibody in phosphate-buffered saline (PBS; Lonza, Verviers, Belgium) together with blocking reagent for 30 min. After washing, the cells were incubated with 200 μL of anti-APC microbeads for 15 min and loaded onto a LS MACS column. W8B2+ cells retained on the column were eluted with 3 mL of PBS containing 0.5 M EDTA and 0.5% bovine serum albumin (BSA; Sigma-Aldrich, Taufkirchen, Germany). In the next step, the cells were stained with the indicated antibody conjugates and used for flow cytometric analysis and fluorescence-activated cell sorting (FACS). For FACSorting, cells were stained with W8B2-APC and CD56-FITC and fractionated into W8B2+CD56− and W8B2+CD56+ populations using a FACSAria cell sorter (Becton Dickinson, Heidelberg, Germany).
CFU-F colonies
Colony forming unit fibroblasts (CFU-F) assays were performed by plating either 1 × 105 unselected or 500–5,000 FACS-selected BM mononuclear cells in T-25 flasks, coated with 0.1% gelatin, containing Knockout™ medium supplemented with 20% knockout serum replacement (Invitrogen, Karlsruhe, Germany), 1 mM
Reagents
Antibody conjugates used in these studies include: MSCA-1-APC, MSCA-1-PE, CD271-APC (Miltenyi Biotec Inc., Bergisch Gladbach, Germany), SSEA-3-PE, SSEA-4-PE, TRA-1-60-PE, TRA-1-81-PE, CD56-FITC (Becton Dickinson).
Antigen identification
The 108 WERI-RB-1 cells were washed 2 times in PBS. Cells were lysed by incubation in SDS-RIPA lysis buffer (50 mM HEPES, pH = 7.4; 1% desoxycholinacid, 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 1 mM EGTA, 100 mM NaF with 1% PMSF, and 0.1% aprotinin) for 5 min at room temperature. Protein solutions were centrifuged at 13,000g for 45 min at 4°C. The supernatants were stored at −20°C until used for separation on SDS-PAGE. The amount of protein in cell lysate was determined in duplicate in a microplate reader using the Bio-Rad Dc protein assay (assay kit and reader from Bio-Rad, München, Germany). Crude lysates and supernatants from phosphatidylinositol-specific phospholipase C (PI-PLC)-treated WERI-RB-1 cells were separated on 10% SDS-PAGE gels and the resulting bands were visualized by silver staining. The bands at 68 kDa were cut and used for fingerprint analysis. In brief, the bands were excised and washed 3 times in a 60:40 solution of 100 mM ammonium bicarbonate (pH 7.8)/100% acetonitrile for 1 h at room temperature. The solution was removed and the gel pieces were vacuum-dried for 25 min in vacuum concentrator (Eppendorf Hamburg, Germany). The gel bands were then rehydrated and reduced with 100 mM DTT at 56°C and treated with iodoacetamide for 20 min at RT. Digestion was performed with 250 ng trypsin overnight at 37°C. Peptides were concentrated and spotted on a gold target, mixed with 2,5-dihydroxybenzoic acid (DHB) solution. Peptide mass maps of tryptic peptides were generated by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) using a Bruker Reflex IV mass spectrometer (Bruker Daltonics, Bremen, Germany). The resulting data were used to perform searches of the Swiss Prot and TrEMBL databases using the program MASCOT (
Reverse transcriptase PCR analysis
Total RNA was isolated from the adherent cells by using RNeasy mini kit (Qiagen, Hilden, Germany) and treated with DNase I (Invitrogen, Karlsruhe, Germany) according to the manufacturer's instructions. One microgram of RNA was reverse-transcribed using the ImProm-II Reverse Transcription system (Promega, Mannheim, Germany) as described by the manufacturer's protocol. The PCR was carried out in a 25-μL reaction mixture containing 1 μL cDNA as template, specific oligonucleotide primer pairs (Table 1), and AmpliTaq DNA Polymerase (Applied Biosystems, Darmstadt, Germany) for 35 cycles at 90°C for 1 min, at 55°C or 60°C for 30 s, and at 72°C for 1 min. Ten microliters of the amplification products were separated by electrophoresis in 2% agarose gels and visualized by ethidium bromide staining.
Primer sequences for TNAP were used as described by Arpornmaeklong et al. [36]. GAPDH primers were used as described by Battula et al. [6].
TNAP, tissue non-specific alkaline phosphatase.
Generation of HEK-293/huTNAP transfectant cell line
Transfection-ready GFP-tagged ORF clone of Homo sapiens alkaline phosphatase, tissue non-specific alkaline phosphatase (TNAP, accession number: NM_000478) was obtained from OriGene, Rockville, MD. HEK-293 cells were selected for the transfection because of their lack of reactivity with antibody W8B2. The 105 cells were transfected according to the manufacturer's protocol in the presence of transfectant MegaTran 1.0 (OriGene, Rockville, MD) in RPMI 1640 medium containing 10% FBS (PAA Laboratories, Pasching, Austria). The transfected cells and mock-transfected cells were grown for additional 2 days in the same medium before culture of cells in selection medium containing neomycin analog G418. After 3 days of culture, cells were stained with antibody W8B2-APC and analyzed by flow cytometry for GFP and MSCA-1 expression.
Immunofluorescence staining for flow cytometry
BM cells were washed twice with PBS containing 1% FBS and 0.01% NaN3 (FACS buffer) and incubated with poly-globin for 15 min on ice to block the nonspecific binding. In the next step, the cells were stained with antibody conjugates W8B2-APC, CD56-FITC, and either SSEA-4 PE, SSEA-3 PE, TRA-1-60-PE, or TRA-1-81-PE for 15 min on ice. After washing, the cells were transferred into Falcon tubes and 1 × 106 cells were analyzed on a FACSCanto flow cytometer (Becton Dickinson, Heidelberg, Germany) using the FCS express software (De Novo Software, Ontario, Canada). For triple staining, BM cells were labeled with W8B2-APC, SSEA-3-PE, and CD56-FITC, gated on the W8B2+ population (R2) and displayed in the plot of SSEA-3 versus CD56.
Treatment of cells with phosphatidylinositol-specific phospholipase C
WERI-RB-1 cells were treated with or without 3 U of PI-PLC from Bacillus cereus (Invitrogen, Karlsruhe, Germany) and incubated for 1 h in PBS at 37°C. Cells were then centrifuged at 10,000g for 3 min. The supernatant was collected and separated on 10% SDS-PAGE. The resulting bands were visualized by silver staining. In addition, treated and untreated cells were stained with W8B2-APC antibody. After washing, cells were analyzed on a FACSCanto flow cytometer.
Alkaline phosphatase assay
MSCA-1+ and MSCA-1− primary bone marrow subsets were sorted onto chamber glass slides (BD Biosciences, Heidelberg, Germany) and assayed for TNAP activity using StemTAG Alkaline Phosphatase Staining Kit (Cell Biolabs, San Diego, CA). Staining was performed according to the manufacturer's instructions. Cells were photographed with a Zeiss Axiovert 200 microscope (Carl Zeiss, Oberkochen, Germany).
Preparation of endometrial single cell suspension
The endometrium was scraped from the myometrium and dissociated into single cell suspensions using enzymatic and mechanical means as previously described [28] using collagenase 3 (2.5 mg/mL; Worthington Biochemical Corp., Lakewood, NJ), DNase I (1 mg/mL; Worthington Biochemical Corp, New Jersey) in 0.1 M PBS (pH 7.4) for 45–60 min, followed by collagenase 2 (4 mg/mL; Worthington Biochemical Corp.) and DNase I in 0.1 M PBS (pH 7.4) for 20–40 min. Red blood cells and dead cells were removed using Ficoll-Paque density gradient separation (GE Healthcare, Uppsala, Sweden) and leukocytes with CD45 magnetic beads (Dynabead, Invitrogen) as described [29].
Flow cytometric analysis of purified human endometrial cells
Freshly isolated endometrial cell and epithelial cell suspensions (5 × 105 cells/mL) were incubated with antibodies against W8B2, IgG1 (isotype control; Dako Cytomation, Glostrup, Denmark), or EpCAM (positive control epithelial cell marker, clone BerEP4; Dako) for 45 min at 4°C. Cells were subsequently incubated with PE-conjugated sheep antimouse IgG (1 μL; Dako) for 30 min at 4°C and resuspended in 5% fetal calf serum (Invitrogen)/PBS for flow cytometric analysis using a MoFlo flow cytometer and Summit software (v4.3, Dako Cytomation Inc., Fort Collins, CO).
Immunohistochemistry of endometrial tissue
Frozen sections of human endometrium (5 μm) were fixed in acetone for 5 min at room temperature (RT). Sections were then sequentially incubated with 0.3% hydrogen peroxide (Orion Laboratories, Welshpool, Australia) and protein block (Dako) for 10 min each at RT. W8B2 (undiluted supernatant) and IgG1 isotype control antibodies were diluted in 0.1% BSA/PBS, and incubated for 1 h at 37°C. An EpCAM reactive antibody (1:20 dilution; Dako) was the positive control. Sections were then incubated with Dako-biotinylated Streptavidin LSAB+ System – HRP Kit for 30 min. Staining was visualized using 3,3′-diaminobenzidine (DAB) tablets and urea peroxidase (both from Sigma, St. Louis, MO) for 5 min at RT. Cells were counterstained with Harris's hematoxylin (Amber Scientific, Midvale, Australia) for 30 s and washed with distilled H2O. Sections were mounted in DPX (BDH, VWR International Ltd., Poole, UK) and examined using a Zeiss, AxioVision Release 4.6 (Axioskop; Carl Zeiss, Oberkochen, Germany).
Results
MSCA-1 is identical to TNAP
Our preliminary data have shown that W8B2-defined antigen is sensitive to phosphoinositol phospholipase C (PI-PLC) treatment. After the cleavage of glycophosphatidylinositol (GPI)-linked proteins, the staining of cells with W8B2 antibody was completely abrogated (Fig. 1A). Therefore, WERI-RB-1 cells were treated with PI-PLC and the resulting supernatant was separated by SDS-PAGE. The separation revealed a prominent band at molecular mass of about 68 kDa resulting from both samples (Fig. 1B). Fingerprint analysis of the cut bands revealed tryptic peptides from alkaline phosphatase. In addition, peptides from human serum albumin (HSA) could be also identified as a minor fraction (data not shown). To confirm the specificity of antibody W8B2 for TNAP, HEK-293 cells were transfected with a PrecisionShuttle pCMV6-AC-GFP Destination Vector (OriGene, Rockville, MD) containing the complete coding sequence of human TNAP. Three days after transfection, ∼30% of transfected HEK-293 cells expressed GFP and reacted with W8B2 antibody (Fig. 1C). Mock-transfected cells showed only baseline reactivity with this antibody (Fig. 1D). These data demonstrate that the W8B2 target MSCA-1 is identical to TNAP. In line with the specific protein expression, reverse transcriptase analysis of TNAP mRNA expression revealed a TNAP-specific band only in HEK-293/huTNAP cells but not in mock-transfected HEK-293 cells (Fig. 1E).

Mesenchymal stem cell antigen-1 (MSCA-1) is identical to tissue non-specific alkaline phosphatase (TNAP). W8B2-reactive WERI-RB1 cells were used for the identification of the MSCA-1 antigen. (
Cell surface TNAP/MSCA-1 in BM cells shows enzymatic activity
We have previously shown that BM-derived MSC can be prospectively isolated using the MSCA-1-reactive antibody W8B2 [11,18]. To study whether alkaline phosphatase expressed on the surface of TNAP/MSCA-1+ cells is functional, BM cells were stained with W8B2-APC and CD56-FITC, fractionated by FACS into TNAP/MSCA-1+ and TNAP/MSCA-1− cells, and analyzed for cell surface TNAP activity (Fig. 2A). As shown in Figure 2B, specific TNAP activity was detected only on the surface of MSCA-1+ but not on TNAP/MSCA-1− BM cells. This demonstrates that antibody W8B2 detects functional TNAP on BM cells.
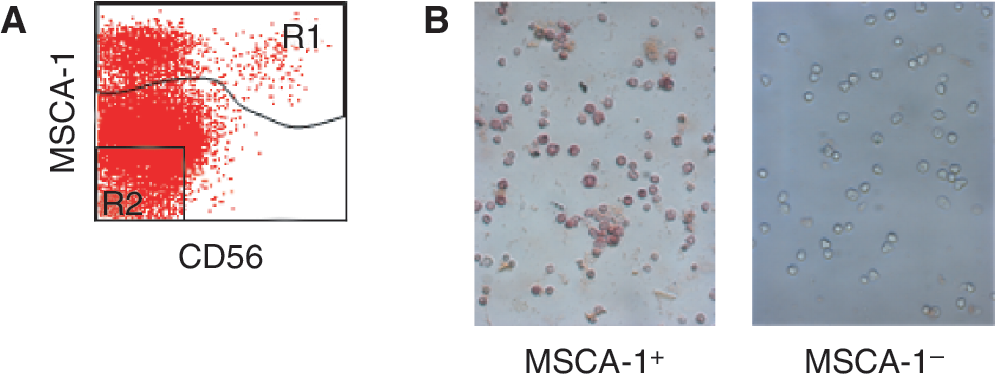
Enzymatic activity of cell surface tissue nonspecific alkaline phosphatase (TNAP)/mesenchymal stem cell antigen-1 (MSCA-1) in bone marrow (BM) cells. BM cells were stained with W8B2-APC and fractionated by FACS into TNAP/MSCA-1+ (R1) and TNAP/MSCA-1− (R2) populations. (
TNAP/MSCA-1+ BM cells express SSEA-3 but not SSEA-4, TRA-1-60, or TRA-1-81
Next, we tested the coexpression of the ES cell markers SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81 on TNAP+ BM cells. As shown in Figure 3, a prominent coexpression of SSEA-3 on the majority of TNAP+ BM cells is detected (79% of TNAP+ cells). However, a minority (21% of TNAP+ cells) were negative for SSEA-3, indicating that SSEA-3 is a novel marker for MSC subsets. In contrast to SSEA-3, the other embryonic markers were negative on TNAP+ cells. To determine whether TNAP/MSCA-1+CD56+ MSC coexpress SSEA-3, BM cells were triple-stained with W8B2-APC, SSEA-3-PE, and CD56-FITC and gated on the TNAP+ population. As shown in the plot of SSEA-3 versus CD56 (Fig. 3B), cells of the rare CD56+ MSC subset do not coexpress SSEA-3.
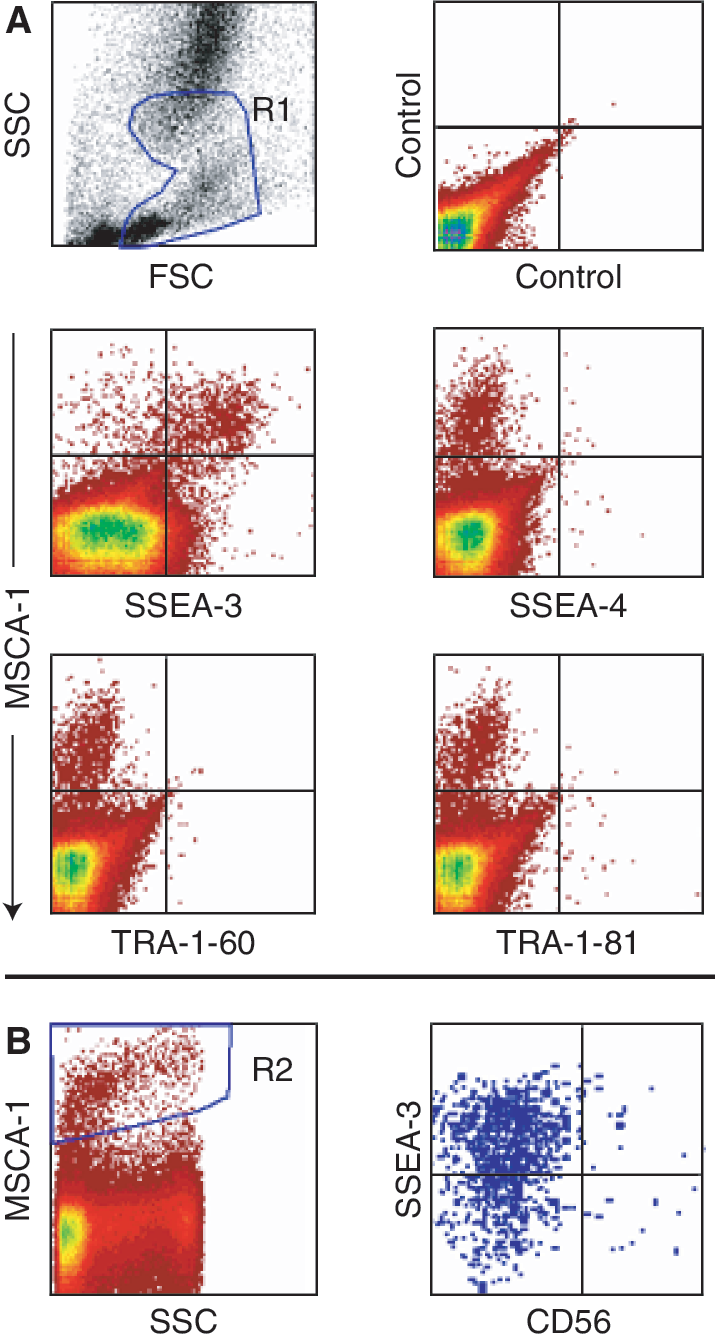
Tissue non-specific alkaline phosphatase (TNAP)/mesenchymal stem cell antigen-1 (MSCA-1)+ bone marrow (BM) cells express SSEA-3 but not SSEA-4, TRA-1-60, or TRA-1-81. (
TNAP/MSCA-1 is differentially expressed on BM cells from different sources
We have recently shown that CD271brightMSCA-1dimCD56+ cells of primary femur-derived BM gave rise to about 2-fold higher CFU-F compared to CD271brightMSCA-1brightCD56− cells [18]. Here, we analyzed the spatial distribution and clonogenic potential of these subsets in different localizations of human bone including acetabulum, trochanter, and trabeculum, and femur. Staining of BM cells with antibodies against CD271, MSCA-1, and CD56 revealed the presence of TNAP/MSCA-1+CD56+ and TNAP/MSCA-1+CD56− subsets, a finding which is in agreement with our previous studies on femur-derived BM cells (Fig. 4A and 4B). Trochanter and trabeculum contained the highest percentage of TNAP/MSCA-1+CD56− cells (71% and 64%, respectively), whereas TNAP/MSCA-1+CD56+ cells were enriched in femur (58%). The level of TNAP expression in the CD56+ population varied considerably: CD56+ cells of acetabulum expressed high levels of TNAP/MSCA-1, whereas trochanter, femur, and trabeculum CD56+ cells expressed this ectoenzyme only at low levels (Fig. 4B). CFU-F assays of 2 separate experiments revealed that acetabulum CD56+ cells did not only express the highest TNAP levels but also give rise to the highest frequency of colonies. In contrast, femur-derived CD56+ cells expressed the lowest TNAP levels and showed the lowest cloning efficiency (Fig. 4C). In agreement with previous results, CD56+ cells of the studied 4 bone sources in all cases gave rise to about twice as many CFU-F as CD56− cells.
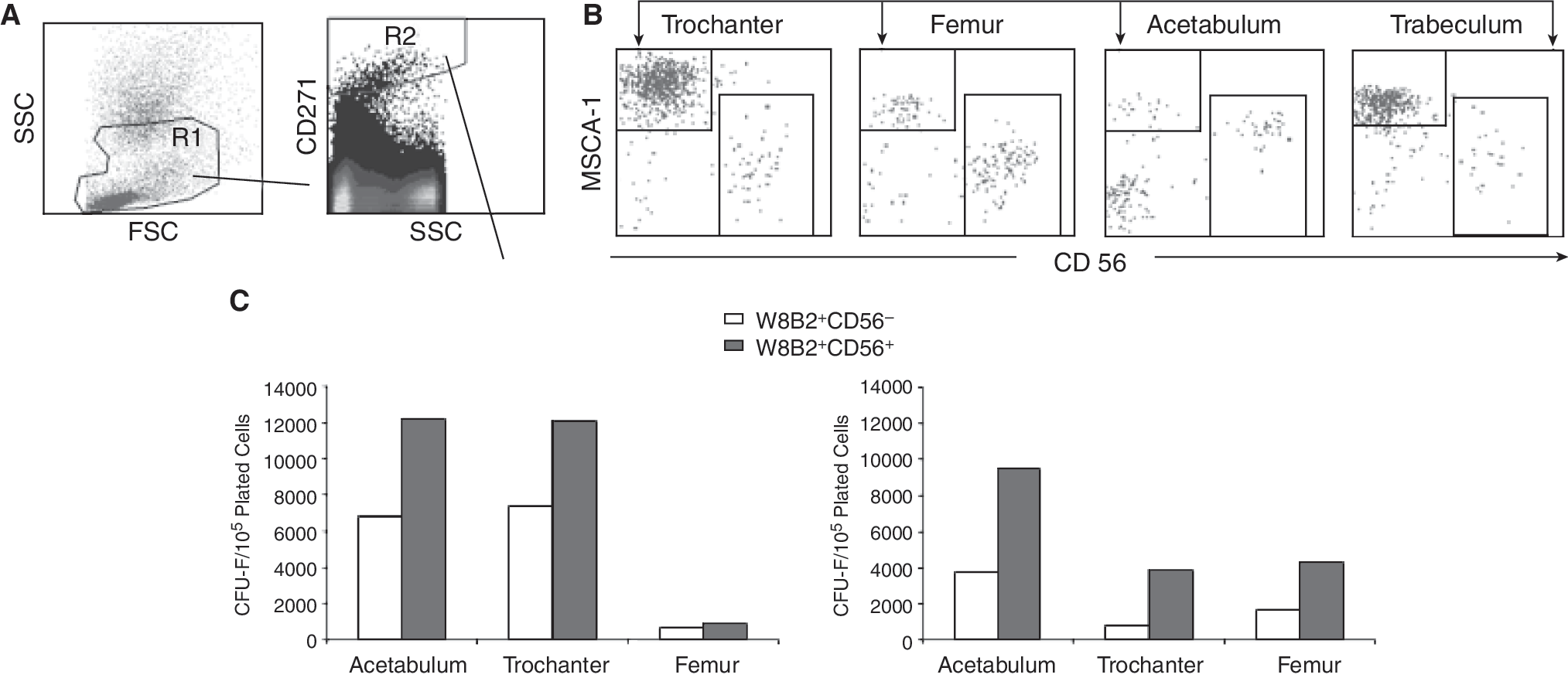
Tissue non-specific alkaline phosphatase (TNAP) is differentially expressed on bone marrow (BM) cells from different sources. Dual scatter gated BM cells (R1) of femur, acetabulum, trabeculum, and trochanter stained with antibodies against CD271-APC, CD56-FITC, and TNAP/mesenchymal stem cell antigen-1 (MSCA-1) were gated on the CD271bright (R2) subset and analyzed for coexpression of CD56 and MSCA-1. (
TNAP/MSCA-1 is expressed on endometrial MSC-like cells and endometrial epithelial cells
Flow cytometric analysis revealed that antibody W8B2 detected a small population of freshly isolated human endometrial cells (Fig. 5A). To investigate whether TNAP is expressed on CD146+ endometrial MSC-like cells [29], cells were double-stained with TNAP-(W8B2) and CD146-reactive antibodies and analyzed by flow cytometry. Figure 5B shows that a small CD146+ population (2.5%) coexpressed TNAP, a similar proportion as documented for CD146+PDGFRβ+ endometrial MSC-like cells [29]. Immunohistochemistry staining showed that TNAP is expressed at high levels on the luminal surface of the glandular epithelium in addition to a weaker expression in blood vessels (Fig. 5C). In agreement with the previously described colocalization of CD146 and PDGFRβ on endometrial MSC-like cells, W8B2 antibody showed also some immunoreactivity adjacent to the vascular lumen composed of pericytes and/or perivascular cells (Fig. 5D).
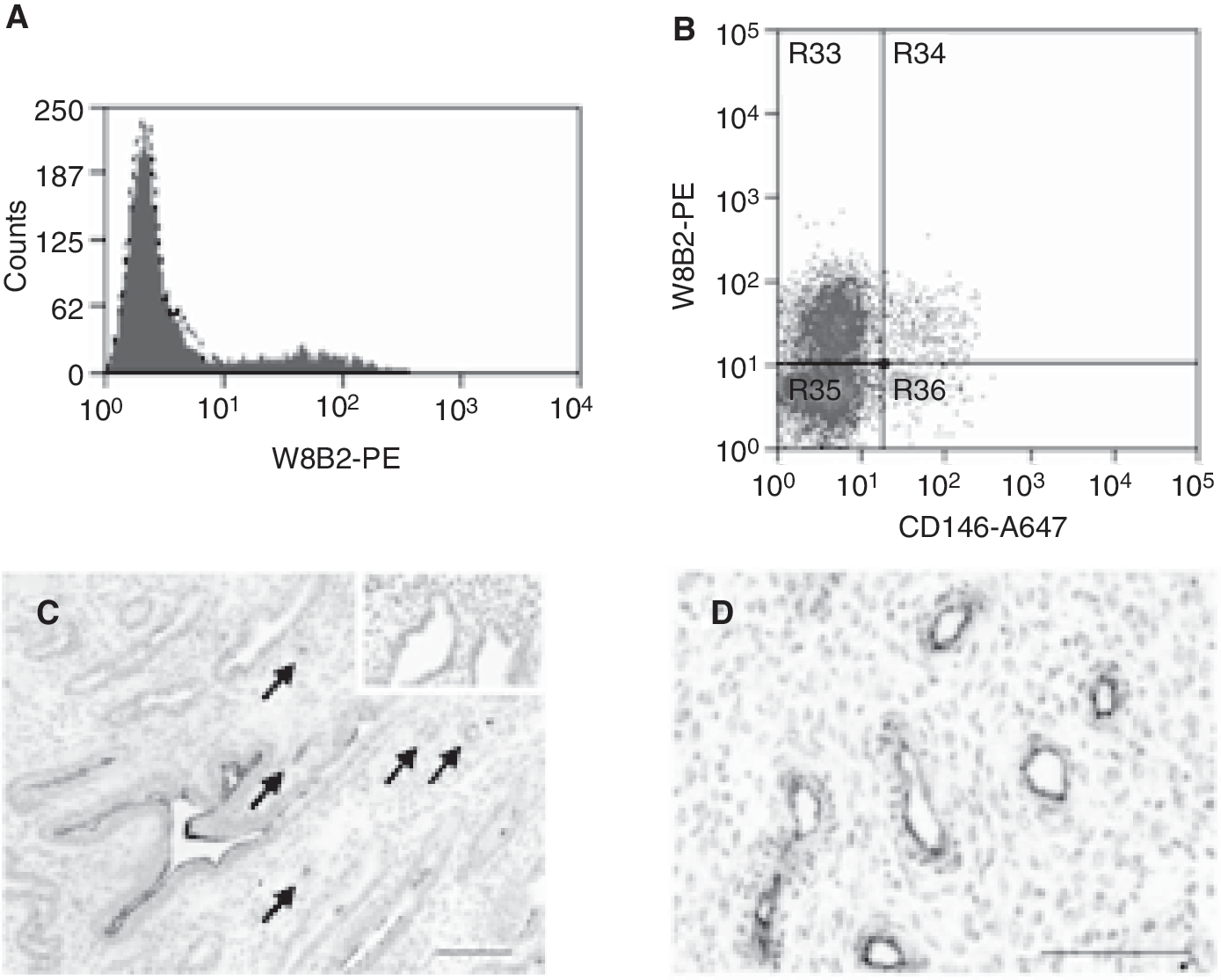
Tissue non-specific alkaline phosphatase (TNAP)/mesenchymal stem cell antigen-1 (MSCA-1) is expressed on endometrial epithelium and mesenchymal stem/stromal cells (MSC)-like cells. (
Discussion
In the present study, we have shown that MSCA-1 is identical to the TNAP. This antigen is a GPI-anchored membrane protein that can be cleaved by treatment with PI-PLC [21,22]. In agreement with this feature, MALDI-TOF MS analysis revealed TNAP sequences in the supernatant of WERI-RB-1 cells treated with PI-PLC. To confirm the identity of TNAP and MSCA-1, HEK-293 cells were transfected with a plasmid containing the complete coding sequence of human TNAP. Flow cytometry analysis showed that MSCA-1-specific antibody W8B2 selectively recognized HEK-293/huTNAP transfectant cells but not the parent cells. In agreement with the TNAP specificity, we could demonstrate that only W8B2+ but not W8B2− bone marrow (BM) cells express the enzymatically active form of the TNAP. In addition, we have shown in different bone sources that only W8B2+ cells gave rise to MSC. In conclusion, we have proved that antibody W8B2 detects functional TNAP on the surface of BM-derived MSC.
TNAP catalyzes the hydrolysis of phosphomonoesters with release of Pi [19,20] and is known to play an important role in bone remodeling by regulating the extracellular concentrations of inorganic pyrophosphate (PPi). PPi suppresses the formation and growth of hydroxyapatite crystals needed for osteoblasts to mineralize the bone matrix [30 –33]. Recently, it was shown that TNAP and low-affinity nerve growth factor receptor (LNGFR; CD271) are up-regulated in mineralizing cells derived from jaw periosteum-derived cells (JPCs). This suggests that TNAP/MSCA-1 and LNGFR are involved in calcification and may serve as a differentiation marker of the mineralizing JPCs [34].
The biological role of TNAP in MSC remains to be clarified. The analysis of the spatial distribution in vivo and determination of in vitro parameters may facilitate to elucidate the physiological function of the TNAP. We observed a negative correlation between the intensity of TNAP expression in the different anatomical sites of the hip joint and the CFU-F frequency. Thus, in acetabulum the level of TNAP expression was lowest whereas the frequency of CFU-F was highest. This is in accordance with the model favored by Gronthos et al. [35] who proposed that early MSC progenitor cells lack TNAP expression but acquire this enzyme during osteogenic differentiation. Our data support the view that the expression of TNAP is restricted to MSC of a later differentiation status. In this respect, it is of interest that the recently identified CD56+ and CD56− MSC subsets show differential expression of TNAP [18]. Thus, the CD56− population with adipocyte differentiation capacity expressed high levels of TNAP whereas the CD56+ subset expressing low levels of this ectoenzyme contained progenitors committed to the chondrocyte lineage. Osteogenic progenitors were found in both fractions. Whether the lower level of TNAP expression in the CD56+ subset is correlated with an earlier maturation status remains to be determined.
In this report, we describe the differential expression of TNAP on CD56+ and CD56− MSC derived from different sites of bone including acetabulum, trochanter, trabeculum, and femur. In the CD56+ subsets, the strongest expression of TNAP was detected in acetabulum, whereas in the CD56−subsets, the highest levels of TNAP expression were recorded in trochanter. Interestingly, the CFU-F were more enriched in acetabulum, independent of the analyzed fraction. Although it is likely that osteoprogenitor cells are enriched in acetabulum because this site contains less marrow and more bone than the other studied sites, specific differentiation assays are necessary to confirm this speculation.
Arpornmaeklong et al. described that a subpopulation of human ES cells induced to differentiate into MSC expresses alkaline phosphatase upon osteogenic lineage commitment. As a consequence, they used TNAP as a key marker for the isolation of ES cell-derived osteoprogenitor cells [36]. Jones et al. used the fibroblast marker D7-FIB as a surface antigen for MSC isolation and reported that BM-derived MSC are positive for alkaline phosphatase enzyme activity [37]. However, they did not show that TNAP is a selection marker for MSC isolation and did not demonstrate TNAP expression on the cell surface. Our report is therefore the first that describes successful isolation of MSC from primary bone marrow tissue.
As the TNAP is expressed at high levels on the surface of ES cells [27], it was of interest to investigate the coexpression of other ES cell markers on TNAP+ BM cells including SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81. Of these, TRA-1-60, TRA-1-81, and SSEA-4 were negative on the TNAP+ BM cells. The fact that SSEA-4 was not expressed on primary BM-derived MSC is in contradiction to the data of Gang et al. who reported SSEA-4 expression on these cells and introduced this molecule as a suitable marker for the prospective isolation of MSC [13]. Most likely, these authors used an antibody from another source. In fact, screening of BM cells with a different antibody claimed to be specific for SSEA-4, stained ∼20% of BM cells. The antibody used in our studies was active since it strongly reacted with ES cells (unpublished). It is therefore likely that SSEA-4 is not expressed on BM-derived MSC. In contrast to the other ES cell-specific markers, SSEA-3 was found to be expressed on the majority of TNAP+ cells (∼80%). Interestingly, the TNAP+CD56+ MSC did not express SSEA-3. Thus, SSEA-3 is a marker that defines a novel subset of BM-derived MSC. Current studies are ongoing to determine the clonogenic and differentiation potential of TNAP+SSEA-3+ and TNAP+SSEA-3− fractions.
In the past, endometrial decidua was reported to express TNAP [25]. Here, we analyzed in more detail the TNAP staining pattern of endometrial tissue and found specific and heterogeneous expression on the luminal surface of glandular epithelia. TNAP is therefore a suitable marker for the isolation of a subset of endometrial glandular epithelial cells. More importantly, TNAP is also expressed on endometrial perivascular cells, where interestingly endometrial MSC-like cells are located [29]. Furthermore, the proportion of W8B2+CD146+ endometrial stromal cells is very similar to the proportion of CD146+PDGFRβ+ MSC-like cells found in human endometrium, suggesting that endometrial MSC-like cells express TNAP and that combined with CD146 this ectoenzyme may be a suitable marker for the isolation from the EpCAM− endometrial stromal cell fraction. The exclusive expression of TNAP in the CD146+ subset, but not on other MSC-like/fibroblast-like cells, suggests that TNAP is developmentally expressed on MSC/pericyte progenitor cells, which is down-regulated during further differentiation.
The precise role of the CD146+PDGFR-β+ subset of endometrium remains to be demonstrated. One possibility is that they are derived from remnant fetal stem cells, which persist after uterine development [38]. These cells have multilineage potential [29] and may originate from the blood vessel wall [39]. Another possibility is that these cells are derived from bone marrow MSC migrating into endometrial tissue during every menstrual cycle [40,41]. The fact that TNAP is expressed in this cell subset suggests that this ectoenzyme plays an important role in the differentiation and endometrial regeneration.
Footnotes
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG), Sonderforschungsbereich SFB-685 (Immunotherapy: Molecular Basis and Clinical Applications) project C10: Development of therapeutic antibodies for the elimination of tumor stem cells, by the DFG project BU 516/2-1: Identification and functional analysis of MSC-specific molecules, and by a research collaboration with TETEC GmbH, Reutlingen, Germany entitled: Isolation and identification of stem cells with adipogenic, chondrogenic, and pancreatic differentiation potential and a National Health and Medical Research Council Career Development Award ID 465121 (CEG) and the Cancer Council Victoria ID 491079 Identifying markers of stem/progenitor cells in normal and malignant endometrium (CEG).
Author Disclosure Statement
No competing financial interests exist.