
Abstract
The goal of this work was to engineer a clinically relevant in vitro model of human prostate stem cells (PSCs) that could be used to interrogate the mechanisms of stem cell control. We, therefore, compared the growth potential of stem cells in 3D culture (where the conditions would favor a quiescent state) with monolayer culture that has previously been demonstrated to induce PSC division. We found a fundamental difference between cultures of primary, adult PSCs grown as monolayers compared to those grown as spheres. The first supported the expansion and maintenance of PSCs from single cells while the latter did not. In an attempt to determine the mechanisms governing stem cell control, several known stem cell activators (including IFNα, FGF2, anti-TGFβ, and dihydrotestosterone) were studied. However, cell division was not observed. CD133+ cells derived from a prostate cell line did not grow as spheres from single cells but did grow from aggregates. We conclude that PSCs can be expanded and maintained in monolayer culture from single cells, but that PSCs are growth quiescent when grown as spheres. It is likely that the physical arrangement of cells in monolayer provides an injury-type response, which can activate stem cells into cycle.
Introduction
M
Human adult prostate stem cells (PSCs) express CD133+ and are restricted to the α2®1 hi integrin population found within the basal epithelial layer [4,5]. In monolayer culture, these cells are highly proliferative, self-renewing, and can reconstitute prostate-like acini in immunocompromised mice [4,5]. Mouse studies have indicated that PSCs are located in the proximal ducts [6], while human studies indicate that they are randomly distributed throughout acini and ducts, often at the base of budding or branching regions [4,5]. These studies indicate that the human adult PSC niche is likely to include interaction with the basement membrane and basal cells.
Investigation of adult human stem cell niches is technically difficult. Generally, there is poor characterization of these niches and only limited cells are available for research. The best studied niche systems are undoubtedly the gonads of Drosophila and Caenorhabditis elegans, where the niche for the germ cells have been defined and can be studied as a whole tissue [7]. These systems have indicated the importance of a defined arrangement of niche cells within 3 dimensions (3D), which is crucial for the maintenance and control of the stem cell. The best characterized adult stem cell systems are the hematopoietic [8] and skin [9], although these are predominantly studied in mouse. The field of adult PSC research has used mouse modeling to great success, as the stem cells can be tracked in situ and micro-dissected to study their properties in vitro [10]. Such studies are not possible in human tissue and for results to be clinically relevant it is essential to study human cells.
To understand the niche, and its control of stem cell fate, new, tissue-engineered models are required to replicate the cells and the extracellular matrix present in the adult human niche. The prostate is a glandular tissue and has been successfully modeled using nonselected cells in 3D to reproduce acini with tissue-like architecture [11]. At present, a similar model has not yet been reproduced from adult human PSCs. Existing models use monolayer culture to expand and maintain PSCs, using type 1 collagen-coated plates, stromal feeder cells, stem cell factor, leukemia inhibitory factor, and GM-CSF [4]. The aim of this work was to combine the existing PSC culture with 3D modeling to recapitulate the adult PSC niche. Our results indicate that in contrast to monolayer culture, in which PSCs can be expanded, sphere-forming assays or semisolid ECM models do not maintain or expand PSCs.
Materials and Methods
Cell line culture
BPH-1 (nontumorigenic prostate epithelia) were kindly provided by Prof. Simon Hayward (Vanderbilt University Medical Center, Nashville, TN) and were routinely grown in RPMI1640 supplemented with 5% FCS (v/v) and 2 mM glutamine. STO (mouse embryonic fibroblasts) cells were routinely grown in DMEM supplemented with 10% FCS (v/v) and 2 mM glutamine.
Tissue collection and isolation of primary cells
Human prostatic tissue was obtained, with consent, from patients (age range 50–90) undergoing transurethral and retropubic prostatectomy for benign prostatic hyperplasia and cystoprostatectomy for bladder cancer. Pathology was confirmed by histological examination of representative fragments. Epithelial and stromal cultures were prepared and characterized as described previously [4]. In brief, prostatic tissue was digested for 12 h with 200 IU/mL of type 1 collagenase (Lorne Laboratories Ltd, Reading, UK) and trypsin, and differential centrifugation was used to enrich for epithelial and stromal fractions. Basal cells were further fractionated on the basis of rapid adhesion to type I collagen. Adherent cells were then sorted for CD133+ and CD133− fractions, using MACS microbeads linked to anti-human CD133, according to the manufacturer’s instruction (Miltenyi Biotec Ltd., Bisley, UK). The purity of the MACS system was calculated as 98% [5]. Overall, freshly isolated cells, from prostate samples, contained 0.395% ± 0.174% CD133+ cells (n = 8), while BPH-1 cultures contained 0.3% ± 0.2% (n = 3).
CD133+ cells were used immediately for experiments or maintained in stem cell media (SCM: keratinocyte serum-free medium with epidermal growth factor, bovine pituitary extract, 2 ng/mL of leukemia inhibitory factor, 1 ng/mL GM-CSF, 2 ng/mL of stem cell factor, 100 ng/mL of cholera toxin) with irradiated (60 Gy) STO cells, added as feeders. Fractionated epithelial cells were routinely cultured on type 1 collagen-coated Petri dishes (BD Biocoat™, VWR, East Grinstead, UK). Due to low cell numbers, individual patient samples were used for each experiment unless otherwise indicated. The stromal cells were routinely cultured in stromal cell growth medium (RPMI1640 supplemented with 10% FCS) and used before passage 3. All cell cultures were routinely cultured without antibiotics in a humidified atmosphere at 37°C and 5% CO2. Bone marrow stroma was cultured as described by Lang et al. [12]. Conditioned media was collected from confluent cells cultures grown for 48 h in stem cell media.
3D semisolid extracellular matrix (ECM) culture
Cells were cultured in SCM and 4% (v/v) growth factor-reduced Matrigel, as described previously [13] or in 1 mg/mL collagen (Becton Dickinson, Oxford, UK), according to the method described in Hall et al. [14]. Cell aggregates were prepared by plating epithelial cells in 50 µL SCM into round-bottomed, 96-well, nonadherent plates (Nalge Nunc, Tokyo, Japan), for 1 week. After this time, 50 µL of 8% (v/v) Matrigel in SCM was added to the existing medium.
For specific experiments, cell were pre-labeled with the red or green fluorescent cell tracker dyes PKH26 and PKH67 (Sigma, St. Louis, MO) according to the manufacturer’s instructions.
3D sphere culture
Primary prostatic epithelial cells, freshly isolated from patients, were plated into 12- or 96-well, nonadherent plates (Nalge Nunc). For experiments using 96-well plates, experimental volumes were 100 µL; for 12-well plates, the volume was 2 mL. The media used was either SCM or neurobasal media (Invitrogen, Carlsbad, CA), supplemented with 2 mM glutamine and B27 (Invitrogen).
Imaging and immunostaining
Immunostaining of cultures was carried out as described previously [13]. In brief, 3D cultures were fixed in 4% (w/v) paraformaldehyde for 20 min (10 min for monolayers), followed by 0.5% Triton X-100 (v/v). Cultures were incubated with primary antibodies for 1 h (1/1,600 or 12.5 µg/mL pan cytokeratin, Sigma; 1/50 or 1.4 µg/mL high-molecular-weight (1, 5, 10, 14) cytokeratins, DAKO; 1/200 or 100 µg/mL CK18-FITC, Sigma; 1/100 or 0.4 µg/mL CD133/2 293C3 Biotin, Miltenyi Biotec Ltd.) followed by 1/500 or 4 µg/mL goat anti-mouse Alexa 568 or 1/100 (20 µg/mL) streptavidin Alexa 568 (Molecular Probes, Invitrogen). Nuclei were counterstained with 0.1 µg/mL DAPI (Sigma) for 5 min.
Apoptotic cells were detected using the Annexin-V-Fluos kit, following the manufacturer’s protocol (Roche, Mannheim, Germany).
Fluorescent cells were imaged using a Multiphoton Zeiss LSM 510 NLO meta-confocal microscope on an Axiovert 200M fluorescent microscope. Phase images were observed with a Nikon TE300 inverted microscope, captured with a Hamamatsu video camera, and analyzed using Velocity 4 (Improvision, Coventry, UK). Digitized images were subsequently prepared using Adobe Photoshop 6.
Flow cytometry
Nonselected cells were stained with PKH67 and a homogeneous subset of PI− PKH67+ cells was isolated using a narrow band gate using a MoFlo cell sorter (Beckman Coulter, High Wycombe, UK). The PI− PKH67max cells were then cultured as indicated above in SCM with STO feeder cells with and without 100 ng/mL Colcemid®. After 4 days the cells were harvested, and the cells stained with 4 µg/mL of anti-CD133-APC (Miltenyi Biotec Ltd., Bisley, UK) and 1 µg/mL of anti Ki-67-Alexa 405 (AbD Serotec, Oxford, UK). Cells cultured in the presence of Colcemid® were used to establish the range of fluorescence exhibited by cells that had not divided during the 4-day post-labeling incubation.
Results
Single CD133+ cells are quiescent when plated in Matrigel
To model the adult PSC niche, it is essential to recreate the architecture of the prostate. Stem cells are in contact with surrounding basal cells, which sit on a basement membrane of matrix proteins in a 3D arrangement. Previously, we have modeled prostatic acini, with tissue-like architecture, using an nonselected population of epithelial cells, derived from both cell lines and primary cultures, in Matrigel [11,15]. Therefore, to determine which phenotype has the potential to regenerate prostatic acini in 3D, CD133+ and CD133− cells were plated into Matrigel, under nondifferentiating conditions (in SCM and in the absence of androgens). Cell plating densities were selected to ensure that aggregation of cells in gel culture could not occur and after plating, single cells were checked to discount the presence of doublets or aggregates. After 10 days in culture, acinus-like spheroids had grown from primary CD133− populations, with a colony-forming efficiency of 2.2% ± 0.75% (n = 8 patient samples), but not from CD133+ cells (n = 5 patient samples). Similarly, single CD133− cells, derived from a prostate cell line BPH-1, grew into spheroids with a colony-forming efficiency of 25% ± 4%. However, spheres did not form from CD133+ cells, derived from BPH-1 (Fig. 1).
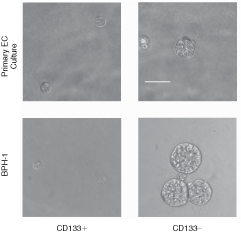
The growth of CD133+ or CD133− selected cells from cultures of primary benign epithelia or BPH-1 cell line, in Matrigel. The 3,000 (primary) or 4,000 (BPH-1) cells/mL were plated (as single cells) and grown for 10 days in 4% (v/v) Matrigel, containing stem cell media (SCM) + 2% FCS. Scale bar = 50 µm.
To ascertain whether the CD133+ cells remained viable as single cells, primary cultures were maintained for a further 8 weeks. At this time point, ∼60% of the CD133+ cells were viable (42% ± 24% of cells were apoptotic or necrotic; results not shown). Despite viability in 3D culture, CD133+ cells did not divide to form acini. As CD133+ cells are routinely expanded in monolayer culture, on type 1 collagen, we plated CD133+ cells into collagen (type 1) gels, but growth was not observed from single cells (results not shown).
Cell division is initiated by the aggregation of CD133+ cells derived from cell lines but not primary cultures
To test if CD133+ cells need cell-to-cell contact to initiate cell division, CD133+ cells were plated at a density to initiate aggregation, for 1 week, after which time they were cultured in Matrigel (as described above), under nondifferentiating conditions. This culture method successfully led to the growth of 3D acinus-like structures from CD133+ cells derived from the BPH-1 cell line (Fig. 2), but not from primary cultures. CD133+ cells from the BPH-1 cell line grew into large budding, acinus-like structures, which contained hollow, luminal structures (Supplementary Fig. 1; Supplementary materials are available online at http://www.liebertpub.com/). Higher magnification images indicated the presence of ductal structures as well as acini (Fig. 2). The aggregates of CD133+ cells, derived from primary cultures, were maintained in culture for up to 8 weeks in Matrigel, but growth was not observed.
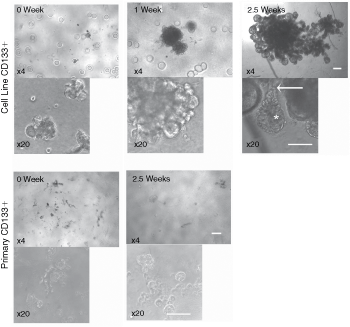
Growth of aggregated CD133+ cells in Matrigel. CD133+ cells were isolated from the BPH-1 cell line or primary prostate epithelial cultures. Three hundred cells were aggregated in 100 µL stem cell media (SCM), for 1 week, in nonadherent 96-well plates (week 0). Aggregates were then grown in 4% (v/v) Matrigel. Cell lines developed acinus-like structures after 1 week and by 2.5 weeks, multiple, hollow acini (asterisk) could be seen at the tips of ductal structures (arrow). Primary cultures showed no further growth (2.5 weeks shown). Four patients’ samples were used in this experiment; 1 was from freshly isolated cells and 3 were primary cultures. Scale bars = 50 µm.
To test whether CD133+ cells (from primary cultures) require physical contact with stromal cells to initiate growth, we aggregated CD133+ or CD133− cells with and without STOs or primary prostate stroma. In the presence and absence of prostate stroma and STOs, CD133+ cells formed an aggregate, but a further increase in size was not observed (Fig. 3), despite culturing for a further 5 weeks (2/2 patient samples). In contrast, using the same patient samples as well as 2 further samples, recombinants of CD133− cells with STOs or CD133− cells alone formed an aggregate and then developed into large epithelial, acinus-like structures. However, recombinants of CD133− cells with primary prostate stroma formed aggregates but did not then develop acinus-like structures. Aggregated STO cells cultured alone in Matrigel do not form smooth-edged acinus-like structures, but remained as aggregates (results not shown).
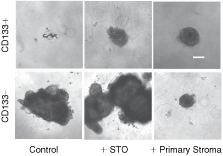
Growth of CD133+ cells and stromal aggregates in Matrigel. CD133+ or CD133− cells were isolated from prostate epithelial cultures. Fifty cells per well were aggregated with or without 100 stromal cells/well (in nonadherent 96-well plates), in a volume of 100 µL stem cell media (SCM) for 1 week. Aggregates were then grown in 4% (v/v) Matrigel for a further 2 weeks, after which times images were collected. Epithelial cells grown with STOs were pre-stained with red cell tracker dye to locate them within aggregates. Fluorescent images were collected at equivalent exposure settings. Images were taken at 4× magnification. Scale bars = 50 µm.
To track epithelial cells within aggregates, CD133+ and CD133− cells were stained with a red fluorescent dye, before aggregation with STOs. After 2 weeks, the CD133+ cells within aggregates remained bright, while CD133− cells were much dimmer (Fig. 3). This indicated that CD133− cells had undergone a number of cell divisions, to form the large acinus-like structures, resulting in dilution of the tracker dye. CD133+ cells had not divided or had divided infrequently. Bone marrow stroma, and stroma derived from prostate cancer both can stimulate prostate cancer growth [16,17]; therefore, they represent potential candidates for stimulating normal stem cell growth. Using 3 different patient samples the recombinant growth of CD133+ cells with each stromal type was tested, but neither led to an increase in the size of aggregates (results not shown). In addition, we placed aggregates of primary CD133+ cells, together with prostate stroma, into collagen 1 gels, although the aggregates were maintained, no further growth occurred (Supplementary Fig. 2).
Aggregation is a feature of prostate sphere-type cultures
We next investigated whether a nonadherent culture model could stimulate PSCs into cycle. Nonselected primary epithelial cells were plated into nonadherent culture plates in stem cell media (SCM) or neurobasal medium. At high plating densities (5,000 cells/cm2), large spheres developed within 5 days in both SCM (Fig. 4A) and neurobasal media (results not shown). By measuring the volume of a spheroid and dividing by the volume of a cell (assumed to be 7 µm3), we were able to estimate that a typical spheroid contained 105 cells. This equates to 17 population doublings, in 120 h, or a doubling time of 7 h. Clearly, such spheres had developed by aggregation of cells and not by cell division. To confirm this, we carried out serial dilutions of nonselected cells. Cell aggregates were observed after only 2 h in culture, at plating densities of 5,000, 1,000 (Fig. 4B), and 100 cells/cm2. Further aggregation of one sphere to another occurred at high plating densities (5,000 cells/cm2), but not at 2,000 cells/cm2 (Fig. 4C). By following the size of the spheres in culture, we observed that spheres plated at 2,000 and 1,000 cells/cm2 increased in size, but cells plated at a density of 100 cells/cm2 (10 replicates) formed aggregates but did not increase in size. This result was confirmed in 3 further primary samples. Disaggregation of spheres into single cells was difficult but careful replating, at equivalent plating concentrations to that of the primary plating density, did not lead to the formation of secondary spheres, after 4 weeks in culture. Indeed, after 2 weeks of secondary culture, we found that 79% ± 15% of cells were annexin V-positive indicating that the majority of cells had undergone apoptosis (results not shown).
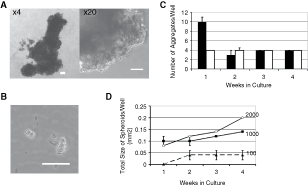
Growth of primary prostate cultures as nonadherent spheres. (
Prostate sphere cultures do not initiate from single cells nor maintain expression of CD133+
To confirm that sphere culture cannot maintain or expand the prostate sphere-initiating cell, we performed limiting dilution experiments on CD133+ and CD133− selected cells from freshly isolated prostate samples (Fig. 5A). CD133+ cells showed moderate growth at a plating density of 100 cells/well (3-fold increase in sphere size). However, the size of spheres did not increase beyond 21 days. At this concentration small, loose aggregates formed in 13/15 wells (Fig. 5B). Further analysis, of annexin V expression, indicated that most cells were apoptotic. Secondary colonies were not observed (Fig. 5A). These experiments were repeated on 3 further patient’s samples. Growth was not observed at plating densities of 50 or 100 cells/well.
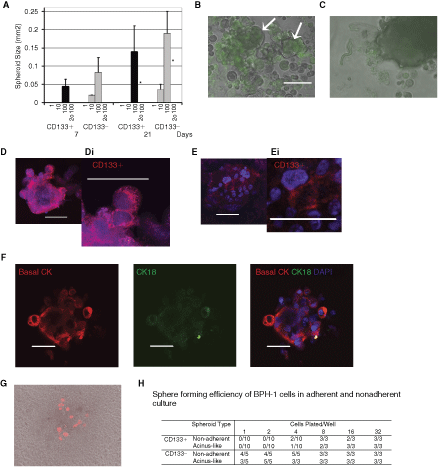
Limiting dilution of CD133+ and CD133− selected prostate cells in sphere culture. (
In contrast, CD133− cells formed large, tight aggregates at a density of 100 cells/well, which grew in size (∼2-fold over 14 days) and showed little apoptosis (Fig. 5A and 5C). Nonetheless, CD133− cells were incapable of secondary colony formation. This result was confirmed in 2 other patients’ samples. Determination of population doublings was difficult to calculate from sphere size because of the low cell numbers and the fact that individual cells were clearly increasing in size (Fig. 5B and 5C). CD133− cells were capable of sphere formation and growth at a density of 10 cells/well, but only from 5 out of 15 wells or 33%. The most significant result was that neither CD133− nor CD133+ cells grew from 1 cell/well. The accuracy of plating 1 cell/well was verified by microscopy (43 replicate wells were confirmed to contain 1 to 3 CD133+ cells/well and 16 wells contained 1 to 3 CD133− cells). This finding was confirmed in 2 separate patients’ samples.
Previous work on prostate epithelial cells has demonstrated that single cells, plated in nonadherent culture, undergo differentiation [18]. To determine whether cells within spheres can maintain CD133 expression, spheres were stained for CD133 after 24 h or 7 days in culture (Fig. 5D and 5E). The use of appropriate CD133 antibodies is very important since human and mouse CD133 is expressed in a broad range of adult epithelial cells, including the stem and progenitor populations [19]. The specificity of the widely used antibody against human CD133 (clone, 293C3) is dependent upon antigen glycosylation, which is only seen on the surface of more primitive cell types [19]. After 24 h in culture, confocal imaging indicated strong expression of CD133 by all the cells in CD133-derived spheres. At higher magnification, expression was localized to the cell surface, particularly at cell-to-cell contacts and within the cytoplasm. After 7 days, CD133 was expressed weakly and only in a minority of cells. Further analysis of 7-day-old spheres indicated that they predominantly expressed basal cytokeratins, while luminal cytokeratin 18 was found weakly in large cells on the sphere surface (Fig. 5F).
CD133+ prostate epithelial stem cells can be expanded in monolayer culture with STO feeder cells [5]. To test whether the lack of growth, observed in 3D culture, could be reversed by culturing in monolayer, CD133+ cells were cultured for 1 week under nonadherent conditions to induce the formation of spheres and were then replated with or without prior disaggregation, in monolayer culture (on type 1 collagen plates). Despite culturing for up to 5 weeks in SCM, colonies were not observed. Of the 50 cells plated, without prior disaggregation (from 4 replicate wells), ∼13–21 cells had survived per initial spheroid but had not subsequently divided (Fig. 5G). When spheres were disaggregated with trypsin and replated as before, only 2 single cells were identified from a total of 4 spheres replated (200 cells in total).
Selected cells derived from the BPH-1 cell line could form spheres in Matrigel; therefore, we tested their ability to form spheres under nonadherent conditions and importantly, if those spheres could form from a single cell (Fig. 5H). CD133+ cells formed spheres, with a sphere-forming efficiency (SFE) of 100% if plated at a density of at least 8 cells/well and those spheres when placed in Matrigel formed acinus-like structures. However, spheroid efficiency dropped to 20% if cells were plated at a density of 4 cells/well and only 1/10 spheres subsequently formed an acinus in Matrigel. Importantly, CD133+ cells plated at a density of 2 and 1 cell/well were unable to form a sphere or acini. We also compared the SFE of CD133− cells. SFE was 100% when plated at a density of 4 cells/well and above, and acini formed with equal efficiency. Unlike the CD133+ cells derived from BPH-1 and primary prostate cells, spheres and acini formed from single CD133− BPH-1 cells with an efficiency of 80% and 60%, respectively.
CD133+ cell division in 3D culture cannot be activated with cytokines or hormones
We have shown that adult prostate CD133+ cells can survive in 3D culture but do not divide. We next attempted to activate the cells into division using a variety of hormones, growth factors, and cytokines known to be important for stem cell cycling and prostate growth. We plated CD133+ cells under nonadherent conditions, with or without STO cells, and grew them in the presence of a test factor, with or without Matrigel. Neutralizing antibodies to TGFβ were investigated as this growth factor maintains dormancy in mouse PSCs [10]. Using increasing concentrations of anti-TGFβ, we found that the growth of CD133+ cells, either grown under nonadherent conditions or when grown in Matrigel, was not affected (Fig. 6). The addition of stroma to the cells also had no effect on growth. Examples of growth at 2 weeks of culture are shown in Figure 6. In addition, we also tested FGF2 because it can support sphere growth in other tissues [20], IFNα because it can activate dormant hematopoietic stem cells (HSCs) [21], and IL-6 because we have previously shown it to be up-regulated in prostate cancer stem and committed cells and therefore represents a candidate for stimulating normal stem cell growth [3]. We investigated the importance of protein methylation, using adenosine peroxidate, which can reverse growth arrest and quiescence as described for B cells [22]. Dihydrotestosterone (DHT) is required for prostate regeneration after castration and acts as a paracrine factor via the stromal cells [23]. Therefore, DHT was used in combination with stromal cells. However, we found no evidence that any of these factors could affect the division of primary CD133+ cells grown in 3D culture (Fig. 6). Each factor was tested on at least 2 primary cultures, and cultures were maintained for up to 12 weeks. In addition, we also cultured spheres in the presence of media conditioned by either STOs or primary prostate stroma, but again growth was not affected.
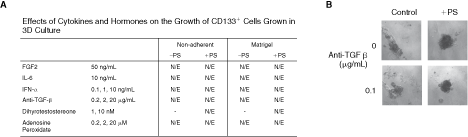
Effect of cytokines and hormones on CD133+ cell growth in 3D culture. (
CD133+ cells undergo cell division in monolayer culture
We have previously demonstrated that prostate epithelial stem cells (CD133+/α2®1 hi) can initiate colonies in monolayer culture and in vivo [4,5]. However, once a colony is found the stem cells stop dividing or divide more slowly relative to the amplifying population because the numbers of CD133+ cells within a culture remain at a low concentration (∼0.1%). To test this, CD133+ cells were isolated from low passage primary cultures (p0–p1) and labeled with the cell tracker dye PKH67. The cells were sorted and subsequently cultured for 4 days and analyzed for CD133, and Ki-67 expression (Fig. 7). We found that >99% of the cell population had divided within 4 days, this included all of the CD133+ population as they had less PKH67 dye compared to the colcemid-treated (nondividing) population (Fig. 7B). The nondivided, PKH67hi population (in R4, Fig. 7B) (0.83%) were not CD133+. A range of PKH67 fluorescent intensity was observed in the CD133+ population indicating different rates of division. However, only a minority (12%) of the CD133+ cells remained in cycle and was Ki-67-positive after 4 days in culture. We verified our findings using a different stem cell marker CD117, which has recently been shown to be expressed by murine prostate epithelial stem cells [24] Although, it is currently not known if CD117 is expressed by human PSCs, we found that the CD117+ population also divided in monolayer (results not shown). To prove that the colonies, in monolayer culture, were initiated from single cells, CD133+ cells were labeled with PKH26 (red) and PKH67 (green) and plated in a 1:1 ratio (Fig. 7D). Doublets were observed within 3 h of plating, but were either red:red or green:green. Only 0.8% of doublets were red:green and those were observed 21 h after plating. This result suggests that colonies founded by CD133+ cells were clonal and were not due to cell migration.
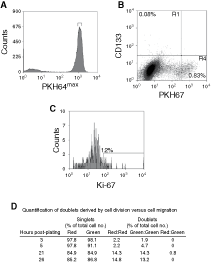
Tracking CD133+ cells in monolayer culture. (
Discussion
There is now accumulating evidence to suggest that diseases, such as cancer, may be derived from the stem cell compartment. To investigate the control of stem cells, within their niche, it is crucial to engineer clinically relevant in vitro models, using adult human tissue. We have found a fundamental difference between cultures of primary, adult PSCs grown as monolayers compared to those grown as 3-dimensional spheres. Monolayer culture supports the expansion and maintenance of CD133+ from single cells [4,5] while 3D culture does not. Growth in 3D, from CD133+ cells, was only observed at high plating densities; however, PSCs were not maintained by this culture system. Our results are consistent with nonadherent embryonic stem (ES) culture, in which differentiation is induced in 3D, whereas pluripotency is maintained when the cells are grown in monolayer with feeders [25].
Both cell line and primary culture-derived CD133+ cells, at clonal density, were noncycling in 3D semisolid matrix culture, but the majority of cells survived under these conditions indicating that provision of a matrix to all surfaces of a PSC maintains survival but inhibits cell division. However, aggregation of CD133+ cells from a cell line (but not primary cells) enabled cell division. Since the environmental signals were identical for both, this indicates that the inherent properties of cell lines are able to overcome quiescence once cell-to-cell contact is initiated. Interestingly, aggregates of CD133+ cells, derived from the BPH-1 cell line, were multipotent and generated both ductal and acinus-like architectures in Matrigel, whereas CD133− cells produced only acinus-like structures and nonselected cells rarely produced ductal structures [11]. This result indicates the multipotent ability of the CD133+ cells.
Examination of the growth of CD133− cells indicated that, at clonal densities, they would only initiate colonies in response to ECM contact and not under nonadherent conditions. These results emphasize the importance of cell-to-matrix contact for prostate progenitor cell survival in 3D. Adhesion of epithelial cells to ECM proteins is mediated by receptors of the integrin family. Experiments with cultured prostate epithelium, keratinocytes, and colonic crypt cells show that differentiation and cell survival is matrix–integrin-dependent [4,26 –28]. The inhibition of β1-integrin signaling causes terminal differentiation of PSCs and amplifying cells [18]. This published data, and the results reported here, emphasize the importance of cell-to-cell contact and the presence of matrix when establishing a niche model for stem cell study.
The hypothetical basis of the sphere assay is that only stem cells, when grown under nonadherent conditions, will survive whereas non stem cells will undergo anoikis [29,30]. However, recent studies from the neural field show that neurospheres are derived from stem and progenitor cells [31,32]. Cell aggregation is also associated with the successful propagation of sphere culture systems. Murine-derived mammospheres have been successfully grown from nonselected cells when plated at high cell densities [29]. Another concern is that secondary and tertiary cultures are often smaller and more differentiated than the parental spheres [29,31 –34]. Apoptosis is also a feature of neurosphere and mammosphere culture and the proportion of stem cells in neurospheres is low in comparison to those in monolayer, adherent culture [33]. It remains debatable whether mammospheres can form from single cells as recent work suggests that aggregation of cells is required to form large mammospheres with limited self-renewing capacity, while single cells form small spheres with unknown self-renewal capacity [35].
The growth of defined populations of mammary and prostate cells has been examined by others, using sphere and semisolid 3D models. In the mammary gland, putative stem cells were defined as CD24med CD49fhi [36] or Lin−CD29hiCD24+ [37]. In both studies, the cells grew in 3D, but these experiments were not carried out at clonal densities. Moreover, such populations are likely to encompass both proliferative and quiescent cells. We previously demonstrated that CD133+ cells from prostate epithelia are largely quiescent [5], whereas the CD133−, but CD49bhi population is actively cycling.
In the mouse prostate, Xin et al. [38] grew clonally derived spheres in Matrigel from nonselected cells, which were capable of self-renewal. Putative stem cells (Lin−Sca1+CD49f+) can also grow in Matrigel when mixed with urogenital sinus mesenchyme [39]. The success of these assays may reside in the use of inductive stroma or more likely, they indicate the difference in the proliferative potential between cells derived from adult mice (10 weeks old) and adult human tissue (50–70 years old).
It remains difficult to measure quiescence and stem cell division in adult tissue. Results from adult mouse HSCs suggest that long-term repopulating cells divide only 5 times in a lifetime [8]. This finding is likely to be true for rapidly dividing tissues (blood, skin, and colon) and that in other tissues adult stem cells may never divide, except in response to injury. Wilson et al. [8] showed that dormant HSCs are not just resting but deeply inactive as they have reduced metabolism, ribosomal biogenesis, and DNA replication. There is further evidence for a continuous decline in stem cell function from birth [40]. The mechanism for this was recently described as the misorientation of centrosomes with age, leading to a reduction in the correct establishment of asymmetric stem cell division with respect to its niche [41].
Provision of stromal and matrix signals, in monolayer culture, clearly provides a different physical arrangement of cells in comparison to culture in 3D. In monolayer, provision of a free surface may act as an injury response to initiate cell division, whereas 3D culture might provide equivalent signals on all cell surfaces. β1-Integrin signals are important for maintaining the architecture of the niche and also for establishing the orientation of cell division [28,42,43]. Asymmetric divisions are important to control maintenance or exit from the niche, and it is likely that monolayer culture provides an exit signal while 3D provides a maintenance signal.
Our experiments highlight the difficulty faced by tissue engineers and cell biologists trying to study the impact of stem cells in adult tissues and disease. There are many limits and technical demands in the use of primary adult culture; availability of tissue, limited cell numbers, and limited self-renewal capacity of the prostate. Therefore, the study of adult stem cells especially from aged tissues requires alternative strategies to model niche systems, to those presented here. In the prostate, monolayer culture provides the most useful model so far for the expansion of adult PSCs. Nonetheless, alternative strategies are still required to study the stem cell niche, such as those used to model skin [44,45]. Organotypic cultures of stroma and keratinocytes in 2D have been developed that are able to maintain label retaining cells for over 10 weeks, reminiscent of vivo-like architecture. Other strategies might include reintroduction of labeled stem cells into organotypic cultures or in vivo [46].
In summary we have shown that normal adult prostate primary stem cells (CD133+) do not initiate sphere formation from single cells or when placed into semisolid extracellular matrix (ECM). Aggregation of CD133+ cells allows limited growth but does not maintain stem cells. In contrast, single CD133+ cells formed colonies, and are maintained at low concentrations, when placed in monolayer culture. The signals that maintain quiescence of normal stem cells are not maintained in cell lines, such as BPH-1. Similarly, CD133− cells can initiate spheres under adherent and nonadherent conditions but importantly, are not capable of self-renewal. The mechanisms controlling stem cell division are intriguing and may be protective to the cell as despite testing a number of key candidates we were unable to initiate cell division of CD133+ cells in 3D culture.
Footnotes
Acknowledgments
We thank Prof Simon Hayward (Vanderbilt University Medical Center, USA) for the BPH-1 cells. We thank Prof. Norman Maitland for his generous provision of laboratory space and members of the Yorkshire Cancer Research Unit for their technical assistance. Many thanks to Mr. M. Stower for providing prostate tissue samples. This work was supported by the Wellcome Trust [GR076612MA] and Yorkshire Cancer Research.
Conference presentations: 8th Annual White Rose Tissue Engineering Meeting, UK, 2006. RegeNer8 Workshop, UK, 2008.