
Abstract
Human embryonic stem (hES) cells hold great promise for application of human cell and tissue replacement therapy. However, the overwhelming majority of currently available hES cell lines have been directly or indirectly exposed to materials containing animal-derived components during their derivation, propagation, and cryopreservation. Unlike feeder-based cultures, which require the simultaneous growth of feeder and stem cells, resulting in mixed cell populations, stem cells grown on feeder-free systems are easily separated from the surface, presenting a pure population of cells for downstream applications. In this study, we have developed a novel method to expand hES cells in xeno-free, feeder-free conditions using 2 different matrices derived from xeno-free human foreskin fibroblasts (XF-HFFs). Using XF-HFF-derived extracellular matrix, together with 100 ng/mL recombinant bFGF-supplemented HEScGRO Basal Medium, long-term xeno-free expansion of hES cells is possible. Resulting hES cells were subjected to stringent tests and were found to maintain ES cell features, including morphology, pluripotency, stable karyotype, and expression of cell surface markers, for at least 20 passages. Xeno-free culturing practices are essential for the translation of basic hES cell research into the clinic. Therefore, the method presented in this study demonstrates that hES cells can be cultured in complete xeno-free conditions without the loss of pluripotency and furthermore, without the possibility of contamination from exogenous sources.
Introduction
S
While it is true that hES cells hold great promise for human cell and tissue replacement therapy, the overwhelming majority of hES cell lines have been directly or indirectly exposed to materials containing animal-derived components during their derivation, propagation, and cryopreservation [19,20]. The presence of animal products in hES cell culture media and feeder cell populations drives 2 main concerns. First, animal products may contain toxic proteins or immunogens that evoke an immune response and thus lead to rejection upon transplantation of the hES cells. For example, hES cells cultured with animal cells or serum products express Neu5Gc, a nonhuman sialic acid that would be immunogenic on cells used for human transplantation [21]. Second, the use of animal products increases the risk of hES cell contamination by the animal pathogens, such as viruses or prions [22]. Hence, the future derivation, propagation, and manipulation of hES cells for the purpose of clinical application should be carried out in xeno-free culture conditions.
There has been some progress in the design of xeno-free culturing practices and components; however, an overwhelming number of studies that claim to be xeno-free actually use products that contain animal or xenobiotic compounds [19,23,24]. In the majority of these xeno-free studies, the main contaminant is basic FGF (bFGF) [23]. bFGF is usually supplied as a recombinant protein derived from bacterial sources. To our knowledge, only Millipore supplies an animal-free version that would be necessary for complete xeno-free culture. In some instances, Knockout™ serum replacer (KRS) is included in xeno-free media compositions even though it contains substantial amount of BSA [24]. However, the most common contaminants including fetal bovine serum (FBS), gelatin (porcine) used for coating dishes, plates and flasks, trypsin–EDTA (porcine) for cell dissociation need to be completely removed from all stages of hESC derivation, maintenance, and differentiation.
Even if animal proteins can be completely removed from hES cell media and culturing products, we are still limited by the substrate required for maintaining cells in an undifferentiated state. While presently feeder fibroblasts have proven the best at supporting hES cell pluripotency and genetic stability, while suppressing spontaneous differentiation [8,25], feeder cells present several complications in hES cell culture, the most important being cellular contamination; furthermore, establishment of clinical grade feeder cell lines for hES cell culture carries with it enormous costs [26]. To circumvent this issue, Matrigel™ has become a popular substrate of choice [27 –31]. Unfortunately, it is derived from murine tumors and is not xeno-free [32]. To address this issue, ECM from human tissue has been used as an alternative in conjunction with several chemically defined culture media [33,34]. Unfortunately in these studies, animal-containing components were still employed. Recently as an alternative, a novel method using poly-d-lysine and the ROCK inhibitor has also been employed [32]. This method requires the continuous application of Y-27632, which leads to gross changes in morphology and as of yet unknown consequences of the hES cells themselves [32]. Most recently, Braam et al. reported that hESCs cultured in mTeSR1 supported hESC growth exclusively on purified vitronectin but not on fibronectin, laminin, and collagen IV [35]. Domogatskaya et al. demonstrated that recombinant laminin-511 could provide a basis for defined surface-coating systems for feeder-free maintenance of undifferentiated mammalian ES cells in vitro [36]. Though these components were not generated in totally xeno-free conditions, these isolated human ECM proteins were found to be effective.
Recently, we derived and expanded human foreskin fibroblasts in completely xeno-free conditions [19]. These cells did not undergo senescence after 55 passages and were able to maintain hES cells in an undifferentiated state. Although these xeno-free feeder cells allowed for the complete xeno-free culture of hES cells, we remained constrained by the need to separate feeders from hES cells for many downstream applications. To circumvent this problem, we investigated whether ECM derived from these xeno-free fibroblasts could be used to support hES cells growth in xeno-free, feeder-free conditions for at least 25 passages. During this long-term culture period hES cells were found to retain typical ES cell features, including morphology, pluripotency, karyotype, and expression of cell surface markers.
Materials and Methods
Xeno-free experimental reagents
Human serum medium (HSM for HFF derivation) and xeno-free medium (XFM for HFF propagation) have previously been described [19]. We used HEScGRO Basal Medium (Chemicon, Temecula, CA), supplemented with recombinant animal-free human fibroblast growth factor-basic (GF003-AF; Chemicon), for human ES cell culture. In addition, we used TrypLE (Animal origin free; Invitrogen, Carlsbad, CA) for HFF and hES cell passaging, as previously described [19].
Preparation of mouse and human fibroblast feeder layers
Mouse fibroblast feeder layers were prepared, as described previously [37]. In brief, mouse embryonic fibroblasts (MEFs) derived from 12.5-day CD1 embryos were treated with 10 µg/mL mitomycin C for 2 h at 37°C. The medium containing mitomycin C was aspirated off, and the cells were washed 6 times with PBS, treated with 0.05% trypsin-53 mM EDTA, centrifuged at 150g, suspended in culture medium, and replated at appropriate concentrations for use in the following week.
Xeno-free human foreskin fibroblasts (XF-HFFs) were derived using a ceiling culture method described previously [19]. Confluent XF-HFFs were mitotically inactivated with 10 µg/mL mitomycin C for 3.5 h, and then dissociated into single cells with TrypLE, counted, and replated on human serum-coated dishes. These feeder dishes were normally used within 1 week.
Preparation of xeno-free conditioned medium (XF-CM) from XF-HFFs
Confluent XF-HFFs cultured on 100-mm dishes were mitotically inactivated with 10 µg/mL mitomycin C for 3.5 h, washed 5 times in PBS and then fed with fresh HEScGRO Basal Medium. After 24 h, the XF-CM was harvested and the XF-HFFs were re-fed with fresh HEScGRO Basal Medium. This process of collecting conditioned media and replacing fresh HEScGRO media can be repeated for up to 7 days with the same inactivated XF-HFFs. Before feeding hES cells, XF-CM was supplemented with an additional 20, 60, and 100 ng/mL of hbFGF, respectively. XF-CM was used fresh or stored frozen at −20°C for 1 month or −80°C for up to 6 months.
Preparation of xeno-free extracellular matrix (XF-ECM) from XF-HFFs
We developed 2 methods for preparing XF-ECM. For both methods, confluent mitomycin C-treated XF-HFFs were incubated for 7 days to allow the build up of extracellular matrix. In the first method (Matrix Chemical Lysis; MCL), cells were rinsed in PBS and incubated in lysis buffer at room temperature for 10 min, rinsed 5 times with PBS, and air-dried in a laminar flow hood. The lysis buffer consists of 0.5% (v/v) of Triton X-100 in PBS and 35 mL of NH4OH per 100 mL of PBS–Triton X-100. In the second method (matrix freeze thaw; MFT), cells in media were stored at −80°C overnight. Upon complete thawing, media together with cellular debris was aspirated after which the remaining XF-ECM was carefully rinsed with PBS, and air-dried in the laminar flow hood. For both methods, air-dried plates were ready to use but could also be sealed in parafilm and stored at −20°C for future use.
hES cell culture
hES cell lines, H9 (WiCell) and CA2 (derived and characterized by Dr. Andras Nagy, Samuel Lunenfeld Research Institute) were initially cultured on MEF feeder cells in KSR culture medium consisting of KO-DMEM supplemented with 20% serum replacement, 1.0% NEAA, 2 mM GlutaMax, 0.1 mM β-mercaptoethanol, 25 units 25 µg/mL penicillin–streptomycin, and 10 ng/mL bFGF. They were passaged 5–6 days using mechanical cutting method or 1 mg/mL of Type IV collagenase (Invitrogen, Carlsbad, CA). hES cell lines, H9 and CA2, were cultured on MEF feeder layers in KSR medium for 40 and 29 passages, respectively, before being transferred to animal-free, feeder-free conditions. High-quality undifferentiated hES cell colonies were selected for this study. Cells were cultured on the 2 XF-ECM preparations using 2 media preparations, HEScGRO Basal Medium and XF-CM, using 3 concentrations of bFGF (Table 1). The transferred hES cells were continuously cultured by splitting the cultures 1:3 using the colony cutting method [19]. Y-27632 (Calbiochem, San Diego, CA) was used were specified at 10 µm during passaging only and was not present during the expansion of the hES cells.
C
Plate efficiency and undifferentiated colonies: + (<25%), ++ (26%–50%), +++ (51%–75%), ++++ (>75%).
Y-27632 (±) means that media supplemented with Y-27632 after passage for 24 h not supplemented with Y-27632 during proliferation.
Abbreviations: HSM, human serum matrix; CM, conditioned medium; NCM, non-conditioned medium; MCL, matrix chemical lysis; MFT, matrix freeze thaw.
Cell number determination
Cell numbers were determined as described previously [38]. In brief, hES cell colonies were dissociated into single cell suspension using 0.05% trypsin/0.53 mM EDTA solution. The cells were enumerated using a hemocytometer and an inverted microscope. The fold expansion for each passage [2–20 every second passage] was calculated as the cell number at the time of harvest divided by inoculated cell number. Statistical analysis on cell expansion was carried out using unpaired Student’s t-test for the comparison of 2 groups with a minimal significance of P < 0.05.
Cytology
hES cells cultured in animal-free conditions were examined for the expression of pluripotency markers, Oct4 (sc5279 Santa Cruz, Santa Cruz, CA), SSEA-3, SSEA-4, SSEA-1, TRA-1-60, and TRA-1-81 (gifts from Dr. Peter Andrews, University of Sheffield), using immunofluorescence, as previously described. Alkaline phosphatase staining was assessed using a commercial kit (Chemicon), as previously described [19]. Finally, karyotype analysis was performed using G-banding as previously described [19].
In vitro and in vivo differentiation
hES cells were spontaneously differentiated as embryoid bodies and examined by for marker gene expression via RT-PCR, as previously described [19]. The resulting cell types were also analyzed by marker immunofluorescence using monoclonal antibodies against β-tubulin III (Sigma T8578, 1:1,000) (ectoderm), smooth muscle actin (Sigma A2547, 1:1,000) (mesoderm), and α-fetoprotein (Sigma A8452, AFP, 1:1,000) (endoderm). Antibody localization was detected using fluorescein (FITC)-conjugated AffiniPure goat anti-mouse IgG (H+L) (1:100, Jackson Immunoresearch Lab, Inc., Baltimore, PA).
hES cells were also subjected to the teratoma formation assay as previously described [19]. Resulting tumors were fixed with 4% paraformaldehyde, embedded in paraffin, sectioned at 10 µm, stained with hematoxylin and eosin, and examined using bright-field microscopy.
Results
Composition of ECM derived from XF-HFFs
Extracellular matrix was prepared from XF-HFFs using both the freeze–thaw (MFT) and chemical lysis (MCL) methods. Following mass spectrometry (LS-MS/MS), the resulting protein readouts were separated on the basis of subcellular localization (Table 2). XF-HFFs and ECM generated using either the MFT or MCL buffer method contained extracellular proteins, intercellular proteins, and proteins that make up the cytoskeleton. Both ECM preparations contained Keratin Type 2, fibronectin, laminin β1, and collagen Type 6 in addition to a number of intracellular proteins including cytoskeleton-associated proteins such as actin, tubulin, and vimentin.
P
Comparison of hES cells cultured in different xeno-free conditions
In order to develop a feeder-free, xeno-free culture method utilizing ECMs derived from XF-HFF, a series of culture combinations consisting of matrix, medium, and growth factors was tested. We found that medium supplemented with Y-27632, as well as XF-CM, dramatically improved the plating efficiency of hES cells. The 2 ECM preparations (MCL and MFT) were compared, whereupon MFT was found to be superior and was used in all subsequent steps (Table 1). For example, MFT together with high concentrations of bFGF effectively supported hES cells in an undifferentiated state (Table 1). In most combinations, between 20% and 80% of the hES cell colonies began to differentiate within 5 passages; however after cells were trained to these new conditions, almost no differentiation was observed following 10 passages. Interestingly, the combinations of MFT + CM + bFGF (60 ng/mL) or MFT + NCM (non-conditioned medium) + Y-27632 + bFGF (60 ng/mL) maintained 80%–90% of hES cell in undifferentiated state up to passages 8 and 10, but extensive differentiation occurred at passages 9 and 11, respectively. However, the combinations of MFT + NCM + Y-27632 + bFGF (100 ng/mL) and MFT + CM + bFGF (100 ng/mL) supported 90% hES cells for >15 passages. Comparing both combinations, we found that the former [MFT + NCM + Y-27632 + bFGF (100 ng/mL)] was a better method than the latter [MFT + CM + bFGF (100 ng/mL)] due to higher cell-attachment efficiency with less preparation time being required (Table 1). Although XF-CM promoted cell-attachment efficiency from about 25% to 75%, it was still lower than plating efficiencies (>90%) obtained in medium supplemented with Y-27632 (Table 1). In addition, preparation of XF-CM is time-consuming, and increases the likelihood of contamination. We therefore passaged the hES cells over 20 times and began characterizing cells after passage 20 under MFT + NCM + Y-27632 + bFGF (100 ng/mL) culture conditions.
Morphology of hES cells cultured under xeno-free conditions
hES cells cultured in traditional culture conditions display a small and round morphology accompanied by a high nucleus-to-cytoplasm ratio, the notable presence of 1 to 3 nucleoli, and a typical spacing between the cells. Under xeno-/feeder-free culture conditions, the morphology of hES cell colonies was similar to that of cell colonies grown in traditional culture conditions. hES cell colonies were relatively circular in shape, and formed a distinctly smooth border between hES cell colonies and the ECM. However, unlike hES cells grown on Matrigel in mTeSR medium, which formed monolayer colonies [3], hES cells-cultured xeno-/feeder-free conditions formed multilayer colonies, with a small percentage of hES cell colonies (∼10%–20%) surrounded by differentiated cells with cubical epithelial-like morphology (Fig. 1).
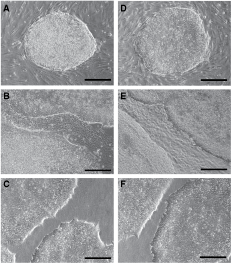
Human embryonic stem (hES) cells morphology grown under different conditions. CA2 (
Proliferation rate of xeno-/feeder-free cultured hES cells
The proliferation potential of hES cells grown in different conditions (MFT + NCM and MEF + KSR) was examined (Table 3). It should be noted that hES cells cultured in MFT + NCM grew at an increased rate compared to hES cells cultured in conventional (BSA-containing medium) conditions. In this experiment, the cell density for replating cultures was ∼3 × 105 per 35 mm culture dish and the cells were passaged every 5 days for MFT + NCM culture, or every 6 days for conventional culture. The average cell expansion was 6.7- and 7.0-folds for CA2 and H9 in MFT + NCM culture compared to 4.8- and 5.1-folds for CA2 and H9 in conventional cultures (Table 3).
C
The cell expansion is the average value of 10 passages. The data were expressed as mean ± SD. Statistical analysis of the data indicated significant difference (t-test, P < 0.05) between matrix MFT + NCM culture and the conventional culture in cell expansion fold of CA2 and H9.
Abbreviations: NCM, non-conditioned medium; MFT, matrix freeze thaw; KSR, Knockout™ serum replacer; MEF, mouse embryonic fibroblast.
Characterization of xeno-/feeder-free hES cells
After 20 passages in xeno-/feeder-free culture conditions, we examined CA2 (Fig. 2A–2F) and H9 (Fig. 2G–2L) hES cells for markers of pluripotency. Both cell lines were assayed for expression of alkaline phosphatase, which was intensely positive throughout each colony (Fig. 2A and 2G). Furthermore, when stained for expression using immunofluorescence, Oct4 (Fig. 2B and 2H), SSEA-3 (Fig. 2C and 2I), SSEA-4 (Fig. 2D and 2J), TRA-1-60 (Fig. 2E and 2K), and TRA-1-81 (Fig. 2F and 2L) all were positive in both cell lines.
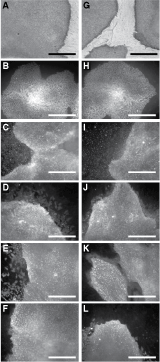
Marker expression in human embryonic stem (hES) cells grown under xeno-/feeder-free conditions. CA2 (
To further characterize the effects of xeno-/feeder-free culture on hES cells, we performed spontaneous in vitro differentiation via an embryoid body (EB) step and analyzed these cells for markers of differentiation using immunofluorescence and RT-PCR (Fig. 3). Cystic EBs were generated from CA2 (Fig. 3A) and H9 (Fig. 3E). After 20 days of differentiation, CA2 (Fig. 3B–3D) and H9 (Fig. 3F–3H) EBs were stained for α-fetoprotein (endoderm), β-tubulin (ectoderm), and smooth muscle actin (mesoderm). Further, we performed RT-PCR on undifferentiated CA2 (Fig. 3I) and H9 (Fig. 3J) cells, in addition to EBs allowed to proceed to 7, 14, or 21 days of in vitro differentiation. By day 21 of in vitro differentiation, each cell line expressed markers from all 3 germ layers (α1-AT, α-FP, enolase, kallikerin, NF-68).

In vitro differentiation of human embryonic stem (hES) cells in xeno-/feeder-free conditions. embryoid bodies (EB) were generated from CA2 (
We also used the teratoma assay as a test of hES cell pluripotency. Upon injecting xeno-/feeder-free cultured CA2 and H9 cells into a SCID mouse, both cell lines generated teratomas consisting of multiple tissue types belonging to all 3 germ layers (Fig. 4, images from H9 teratomas, CA2 images are not shown).
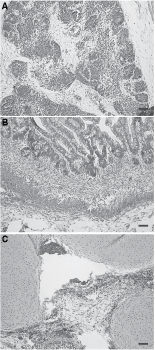
In vivo differentiation of human embryonic stem (hES) cells in xeno-/feeder-free conditions. Teratomas were generated from CA2 and H9 cells after long-term culture in xeno-/feeder-free culture. Representative images are providing depicting pigmented neural epithelium (ectoderm,
Finally, we characterized the xeno-/feeder-free cultured cells for genetic stability using karyotype (Fig. 5). hES cells are very sensitive to chromosome aberrations including the gain/loss of genetic material. However, we found that under xeno-/feeder-free culture conditions both CA2 (Fig. 5A) and H9 (Fig. 5B) maintained a correct chromosomal complement after 20 passages.
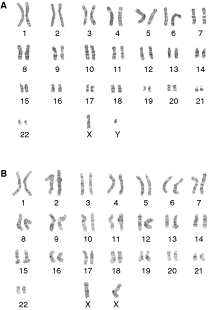
Karyotype of cells after long-term xeno-/feeder-free culture. Both cell lines retained a normal karyotype after extended xeno-/feeder-free culture (>20 passages). CA2 (
Discussion
When contemplating the use of hES cells in clinical settings, it is important to demonstrate that these cells can be both cultured and differentiated under xenobiotic-free conditions. In this study, we found that XF-HFF-derived ECM can support the long-term xeno-free maintenance of hES cells in an undifferentiated state. Moreover, when cells were cultured for prolonged periods in xeno-/feeder-free conditions, they retained a normal karyotype and their ability to differentiate into all 3 germ layers. It was necessary to select only high-quality hES cell colonies for this study, as we discovered that partially differentiated colonies could not be adapted to the xeno-/feeder-free conditions, presumably because they had already lost their pluripotency.
In recent years, many groups have reported the ability to maintain existing hES cell lines as well as derive new lines on Matrigel or extracellular matrix (ECM) prepared from MEFs [39] or human source feeder cells [40 –45]. However, these feeder cells of human origin were not derived or cultured in totally xeno-free conditions. During this procedure, any animal component containing material must be avoided. As a prelude to this work, we developed a xeno-free method to derive fibroblasts from human foreskin tissue [19]. Four XF-HFF lines (I, II, III, and IV) were derived from 4 different foreskins. To date, cell lines I and II have been cultured on human serum matrix in xeno-free medium up to 55 and 47 passages, respectively.
In this study, we derived ECM from mitotically inactivated XF-HFFs using both chemical lysis (MCL) and freeze–thaw (MFT) methods. We observed that MFT was better than MCL in supporting the undifferentiated growth of hES cells. Although both MFT and MCL retained similar overall proteomic profiles compared to that of intact XF-HFFs, the profile of intercellular and cytoskeletal proteins were different between the 2 methods. Therefore, it is possible that the lysis buffer method resulted in the extraction/destruction of a protein that aids in the attachment/maintenance of the hES cells that was present in the freeze–thaw method.
Others have reported that matrix derived from human serum could also support undifferentiated growth of hES cells [46]. However, using their methods we observed that >50% of hES cell colonies differentiated within 5 passages when cultured in medium supplemented 100 ng/mL bFGF. We do concede, however, that numerous inconsistencies are found between batches of human serum, which can possibly explain their results. This reported batch-to-batch variability of human serum has presented difficulties in using human serum for the derivation and maintenance of hES cell lines [47] and highlights the need for alternative approaches to xeno-free culture as presented here.
Aside from the ECM, the culture media must also be free of any animal components. In this study, we used XF-CM prepared from XF-HFFs, which promote cell attachment and help maintain cells in an undifferentiated state. When we compared the use of XF-CM to that of Y-27632, we found similar maintenance and attachment. However, since preparation of CM is time-consuming and can also suffer from batch-to-batch variation depending on the health of the feeder cell population, we chose to forego the use of XF-CM in lieu of Y-27632. Similar to other groups, we found that the feeder-free culture required high levels of bFGF to maintain hES cells in an undifferentiated state. This requirement was not alleviated by the use of XF-CM. Nonetheless, it is interesting to note that there is a factor(s) in conditioned medium, which is as bioactive as Y-27632 in supporting hES cell vitality. In xeno-/feeder-free conditions we observed that the ROCK inhibitor, Y-27632, increased cell attachment after replating, much like in conventional culture conditions.
In conclusion, xeno-/feeder-free culture, as described here, should significantly facilitate the use of hES cells for therapeutic applications. However, we concede that both hES cell lines we used are xeno-contaminated and their propagation in animal-free conditions cannot completely eliminate the exogenous and immunogenic factors in culture [21]. Use in clinical applications would require starting from scratch with newly derived hES cell lines that have never been exposed to animal products under GMP conditions. Despite the ability of these xeno-free conditions to support hES cell maintenance, line derivation has not yet been tested. The results of this study only suggest this possibility. Recently, we have derived and characterized 2 hES cell lines using mechanical dissection of the ICMs (manuscript in preparation). We believe that this new technique, coupled with the results from this study, will aid in the future generation of completely xeno-free-derived hES cell lines.