
Abstract
We have recently demonstrated that peripheral blood monocytes can be differentiated in vitro into hepatocyte-like cells using appropriate differentiation media. Phenotype conversion required prior in vitro culture in the presence of M-CSF, IL-3, and human serum, during which the cells acquired a state of plasticity, so were termed “programmable cells of monocytic origin” (PCMO). Here, we have further characterized the process of PCMO generation with respect to markers of monocyte-to-macrophage transition and pluripotency. During a 6-day culture period, various monocyte/macrophage differentiation markers were down-regulated being indicative of a process of partial dedifferentiation. Dedifferentiation and hepatic redifferentiation also proceeded in highly purified monocyte preparations, albeit with different kinetics, suggesting that the presence of nonmonocytes, or soluble factors derived from them, is not essential in order for monocytes to acquire a multipotent state. PCMOs expressed various markers of human embryonic stem cells with early induction of NANOG and OCT4. Expression of the pluripotency-associated OCT4A isoform was paralleled by a global rise in histone H3 methylation on Lys-4, a marker of active chromatin, and coincided with peak sensitivity to tissue-specific differentiation. These results show that peripheral blood monocytes can be induced in vitro to transiently acquire stem cell-like properties and concomitantly a state of increased differentiation potential toward the hepatocytic phenotype.
Introduction
A
Recently, we have developed a protocol to induce from monocytes by in vitro culture an apparently more plastic derivative, which we named “programmable cells of monocytic origin” (PCMO). These cells following a 6-day treatment with M-CSF, IL-3, and human serum (designated “PCMO culture”) could be induced upon exposure to appropriate induction media to differentiate into cells with certain endothelial characteristics [3], chondrocytes [4], and insulin-expressing cells [5]. The latter cells up-regulate not only the insulin and glucagon genes but also transcription factors involved in pancreatic β-cell differentiation [5]. Recently, we focused our interest on PCMO-derived hepatocyte-like cells (NeoHeps), which express a variety of hepatocyte markers and exhibit hepatocyte-specific metabolic functions [5 –7], making these cells an attractive alternative to primary human hepatocytes for studying drug metabolism in vitro [7]. Moreover, NeoHeps improved survival in a rat model of acute liver failure [8] and monocyte-derived hepatocyte-like cells even showed promise in the treatment of HBV-related decompensated liver cirrhosis [3,9]. The crucial point, from a bio(techno)logical perspective, is that monocytes are capable of coordinated expression of the gene sets required for these nonhematopoietic cell-like activities to occur.
Various mechanisms have been implicated in the acquisition of plasticity/enhanced differentiation potential of adult somatic cells including myelomonocytic cells, such as trans-differentiation, dedifferentiation, or cell fusion [10]. During dedifferentiation, cells silence tissue/cell-type-specific genes and eventually reacquire more primitive features, such as mitotic activity and expression of markers of self-renewal and multi/pluripotency. This would be consistent with previous observations that PCMOs down-regulated PRDMI, the human homolog of murine BLIMP-1, and ICSBP, 2 genes encoding monopoietic transcription factors [5]. However, no information is yet available for other nuclear factors of monocyte → macrophage differentiation (eg, Klf4) or markers associated with specialized functions, for example those involved in sensing and killing of invading microorganisms such as Toll-like receptors (TLRs) and NAD(P)H oxidase, respectively. A more general loss of the monocyte/macrophage phenotype would be in favor of a dedifferentiation process and would lend support to our contention that PCMOs represent cells that have reverted to a more primitive progenitor with less restricted differentiation potential. Hence, in the first part of this study we examine whether altering specific parameters, such as duration of PCMO culture or cellular composition of the monocyte preparation, impact on acquisition of the multipotent state by PCMOs.
Despite the biological importance of the broad differentiation potential of monocytes, surprisingly little is known about the mechanisms that allow them to maintain an uncommitted precursor state. It is reasonable, then, to question whether the mechanisms underlying the phenotypic plasticity of embryonic and adult stem cells might also operate in monocytes subjected to PCMO culture conditions. In this respect, we provide evidence that PCMOs share in common several markers of ESCs such as OCT4 (also termed POU5f) and NANOG, both of which can function as activators of self-renewal and pluripotency genes and as repressors of lineage commitment genes. Specifically, we demonstrate that PCMOs reactivate the endogenous OCT4 and NANOG genes and undergo histone modifications known to be associated with their transcriptional activation. These genetic and biochemical events correlated well with respect to timing and duration with the transiently increased differentiation potential of the cells. We surmise that the generation of PCMOs involves both partial dedifferentiation and the acquisition of stem cell-like properties, providing a molecular explanation for the previously observed phenotypic conversion of peripheral blood monocytes into hepatocyte-like cells.
Materials and Methods
Isolation, purification, and in vitro culture of monocytes
PCMOs were generated from human peripheral blood monocytes following the protocol of Ruhnke and colleagues [5]. In brief, mononuclear cells from either heparinized blood, buffy coats, or LRS chambers were isolated by density gradient centrifugation (Ficoll-Paque; Amersham Pharmacia Biotech AB, Uppsala, Sweden). Cells were cultured in either 24-well or 6-well plates (Cell+, Sarstedt, Nümbrecht, Germany) for various lengths of time in PCMO medium (RPMI 1640 medium (Invitrogen, Karlsruhe, Germany), supplemented with 5 ng/mL final concentration of M-CSF and 0.4 ng/mL final concentration of IL-3 (both from R&D Systems, Wiesbaden, Germany), 90 μM 2-mercaptoethanol, and 10% human AB serum (Lonza, Verbier, Belgium). One hour after plating, cultures were gently washed to enrich for adherent cells and fresh medium was added to the adherent cell layer resulting in enrichment of 60%–70% CD14+ cells. These cultures are referred to as Ficoll-Paque density gradient centrifugation (FC)-purified. Alternatively, monocytes were purified by elutriation as described [11], based on a previously published method [12], resulting in a purity of 95%–97% CD14+ monocytes. Purified cells were cultured for various times (1 h, 2, 4, or 6 days) in PCMO medium, which was replaced in all cultures every other day. Subsequently, hepatocytic differentiation was initiated by replacing the PCMO medium with RPMI medium containing 10% fetal calf serum (Lonza), 3 ng/mL final concentration of FGF-4, and 20 ng/mL final concentration of hepatocyte growth factor (HGF; both from R&D Systems). The addition of HGF, which was not part of the original medium formulation [5,6], further enhanced albumin and Factor VIII expression (A.N. and H.U., unpublished data).
Flow cytometry analysis
Flow cytometry analysis of freshly isolated and cultured monocyte preparations was carried out as described previously [5] with minor modifications. The following antibodies were employed: APC-conjugated mouse antihuman CD14 (clone M5E2; BD Biosciences, San Jose, CA), fluorescein isothiocyanate (FITC)-conjugated antihuman TLR2 (clone TL2.1), and phycoerythrin (PE)-conjugated antihuman TLR4 (clone HTA125, both from eBioscience via NatuTec, Frankfurt, Germany). Only viable cells negative for 7-amino-actinomycin D (7-AAD) were analyzed.
RNA isolation and RT-PCR
RNA was isolated with PeqGold (peqLab, Heidelberg, Germany) according to the manufacturer's protocol. Due to the lack of introns in both OCT4 and NANOG and the presence of several pseudogenes in the human genome, total monocyte RNA preparations were always treated with DNase I (Invitrogen) prior to reverse transcription. Primers were generally chosen to span exon–intron boundaries and, in the case of NANOG, to be unable to recognize the mRNA sequences encoded by the 3 expressed pseudogenes [13]. As positive control, we employed total RNA isolated from the human ESC line SA002.5 (kindly donated by Dr. P. Björquist, CellArtis, Stockholm, Sweden). Standard end-point PCR was performed on a My-Cycler (Bio-Rad, München, Germany) with Taq polymerase and supplied buffers (Invitrogen) and a hot start-touch-down program described earlier [6]. In brief, primers and cDNA template were premixed in a volume of 2.5 μL and added to the remaining 22.5 μL of PCR preheated to 80°C. Denaturation, annealing, and extension were performed at 30 s each. The initial annealing temperature of 61°C was lowered by 2°C every second cycle until a temperature of 51°C was reached at which 30 cycles were performed. PCR products were analyzed by electrophoresis through 2% agarose gels and viewed under UV light after ethidium bromide staining. Quantitative real-time RT-PCR analysis (qPCR) was carried out on an I-Cycler with iQ SYBR Green Supermix (Bio-Rad). Denaturation, annealing, and extension were performed at 30 s each. Forward and reverse primers were designed to have approximately the same T m and annealing temperatures were chosen to be T m −5°C. Calculation of results was performed with the ΔΔCT method following normalization of the data from the gene of interest with equivalent data for the 2 housekeeping genes, TATA box-binding protein (TBP) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). For oligonucleotides used as PCR primers (see Table 1).The primer sequences for ICSBP, PRDMIβ, PU-1, GAPDH [5] and Nox1–5, p22phox, p47phox, and p67phox [14] have been given earlier.
Isolation of nuclear protein, immunoblotting, and immunoprecipitation
Preparation of total cellular lysates, SDS-PAGE, and immunoblotting were carried out as described in detail elsewhere [15]. Nuclear proteins from PCMOs, F9 murine embryonal carcinoma, and JEG-3 human choriocarcinoma cells were isolated using a commercially available kit (Thermo Scientific, Rockford, IL) and protein concentrations were determined with the Bradford assay (Thermo Scientific). Proteins were fractionated by SDS-PAGE, blotted onto PVDF membrane, and probed with the following antibodies: p47phox (Santa Cruz Biotechnology, Santa Cruz, CA), c-Src (GD11; Upstate Biotechnology, Waltham, MA), β-actin, α-tubulin (both from Sigma, St. Louis, MO), E-cadherin (BD Biosciences), Oct4 (Santa Cruz Biotechnology, # sc-5279), Nanog (Abcam, Cambridge, UK), histone H3 (trimethyl K4), histone H3, and histone H4 (all from Biozol, Eching, Germany). Oct4 and heat-shock protein (HSP) 90 were also immunoprecipitated from PCMO total protein lysates with monoclonal Oct4 and HSP90α/β (H-114) antibodies, respectively, and Protein A/G-Plus Agarose (all from Santa Cruz Biotechnology) according to the protocol of the supplier. Following SDS-PAGE and blotting, immunoprecipitated OCT4 and HSP90 proteins in PCMOs and in F9 cells (purchased as positive control lysate from Santa Cruz Biotechnology) were detected using the above mentioned antibodies.
Statistics
Data from qPCR experiments represent the mean ± SD after normalization with the housekeeping genes GAPDH and TBP and were generally derived from 3 wells processed in parallel. Statistical significance was calculated using the unpaired Student's t-test. Data were considered significant at P < 0.05.
Results
The monocyte–PCMO transition is characterized by changes in marker expression for monocyte/macrophage function and cell adhesion
We have previously observed that in response to our specific culture conditions monocytes silence the monopoietic marker genes PRDMI and ICSBP [5]. To reveal whether down-regulation of these genes was part of a more general process of dedifferentiation, we monitored various proteins involved in the sensing and killing of bacteria by monocytes/macrophages, such as CD14, Toll-like receptors (TLRs) 2 and 4, as well as NOX4 and p47phox both subunits of the reactive oxygen-producing enzyme NAD(P)H oxidase. Flow cytometry analysis of cell surface expression of CD14, TLR2, and TLR4 in freshly isolated monocytes and PCMOs of various stages of maturity (n = 4) indicated that the percentage of CD14+ cells decreased to 46.6% (range: 30.3%–61.2%) on Day 6 of culture, while the corresponding values for TLR2 and TLR4 were 4.7% (range: 1.6%–8.4%) and 14.1% (range: 4.6%–26.6%), respectively (Fig. 1A, see also means ± SD for each donor). A time-course analysis revealed that the decrease in CD14 and TLR2 expression commenced between Days 1 and 4 of culture while that of TLR4 was much more rapid and was virtually complete after 1 day (Fig. 1A). Other markers (CD86, HLA-DR) remained constant over the 6-day culture period (data not shown). To evaluate whether down-regulation of the TLRs is controlled at the transcriptional level, we employed qPCR analysis. Interestingly, expression of TLR2 (Fig. 1B), TLR4 (see Fig. 4A), 2 other TLRs, TLR7 (recognizing viral RNA) and TLR9 (recognizing bacterial DNA), and NOX4 (Fig. 1B) was dramatically decreased on Day 6 of culture. In contrast, expression of p47phox remained unaltered at the mRNA level (data not shown), but appeared to be down-regulated at the protein level starting from Day 2 in PCMO medium (Fig. 1C), consistent with earlier reports that p47phox expression is primarily controlled posttranslationally [16]. The switch from suspended to adherent growth, which was associated with changes in cell morphology [5], might also affect proteins involved in cell adhesion and cytoskeletal regulation. In fact, we observed a steady increase in p60Src, β-actin, and E-cadherin (Fig. 1C), while α-tubulin remained fairly constant over the 6-day culture period (Fig. 1C). Together, these data suggest that monocytes during their conversion to PCMOs undergo partial dedifferentiation.
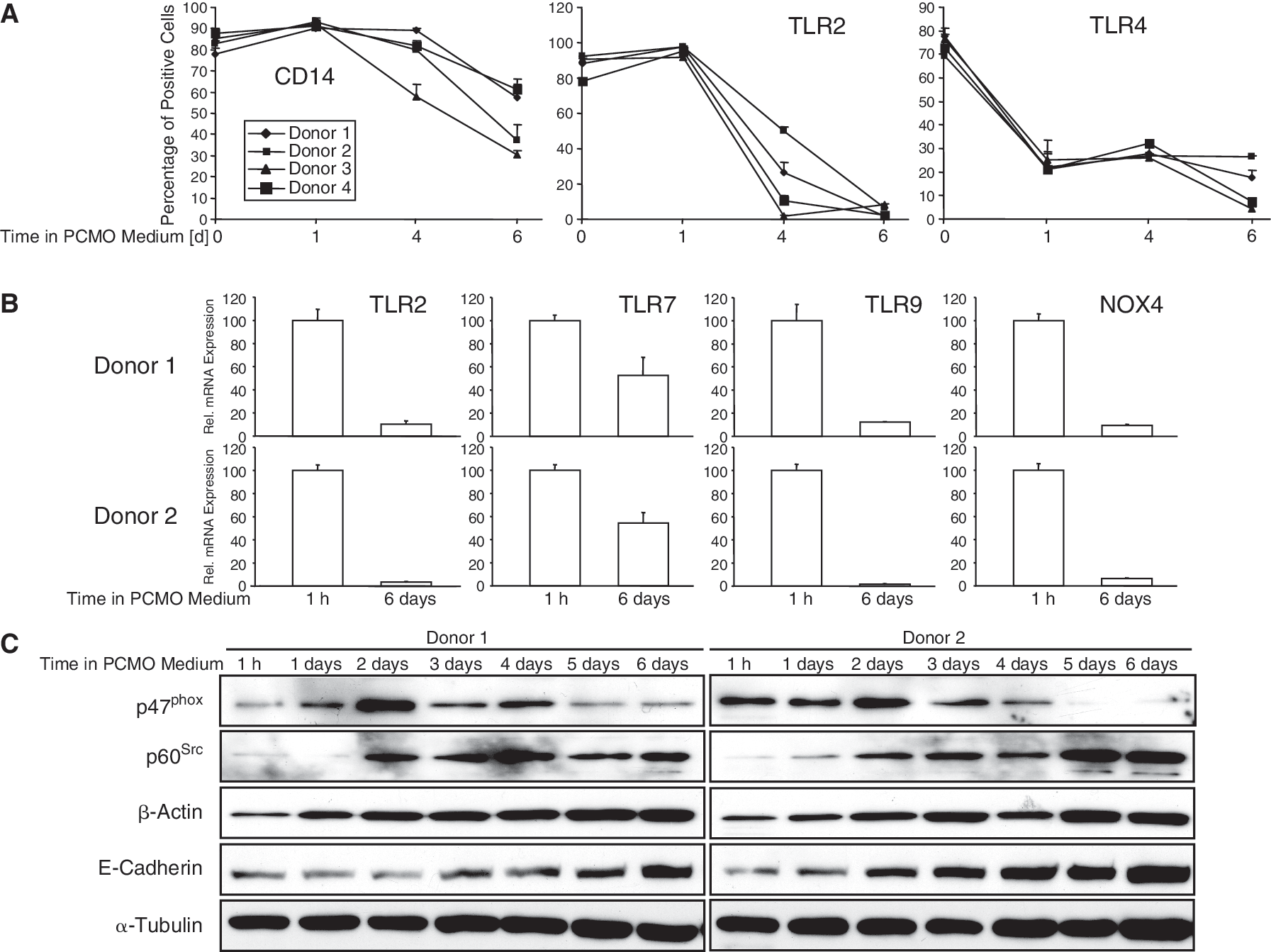
Analysis of monocyte/macrophage markers during programmable cells of monocytic origin (PCMO) culture. Peripheral blood monocytes prepared by Ficoll-Paque density gradient centrifugation (FC) were allowed to adhere for 1 h, or cultured for the indicated times, in PCMO medium prior to harvest. (
Optimal hepatocytic differentiation of PCMOs varies with time in culture
In vitro studies with mouse monocytes have shown that the intrinsic sensitivity toward exogenous differentiation clues to a specific phenotype varies with time in culture, with the differentiation potential to macrophages being the default state [17]. To test whether this also applies to human monocytes, we cultured FC-purified monocytes for up to 6 days in PCMO medium, followed by a 14-day exposure to hepatocyte differentiation medium (Fig. 2A) and monitored the expression of hepatocyte-defining markers as earlier. As shown in Figure 2B, the susceptibility of the cells to hepatocytic differentiation indeed was time-dependent with all 3 markers displaying the highest expression when the cells were kept for 4 days rather than for 6 days in PCMO medium. From these data, we conclude that the susceptibility of cultured monocytes to tissue-specific differentiation fluctuates with time in culture.
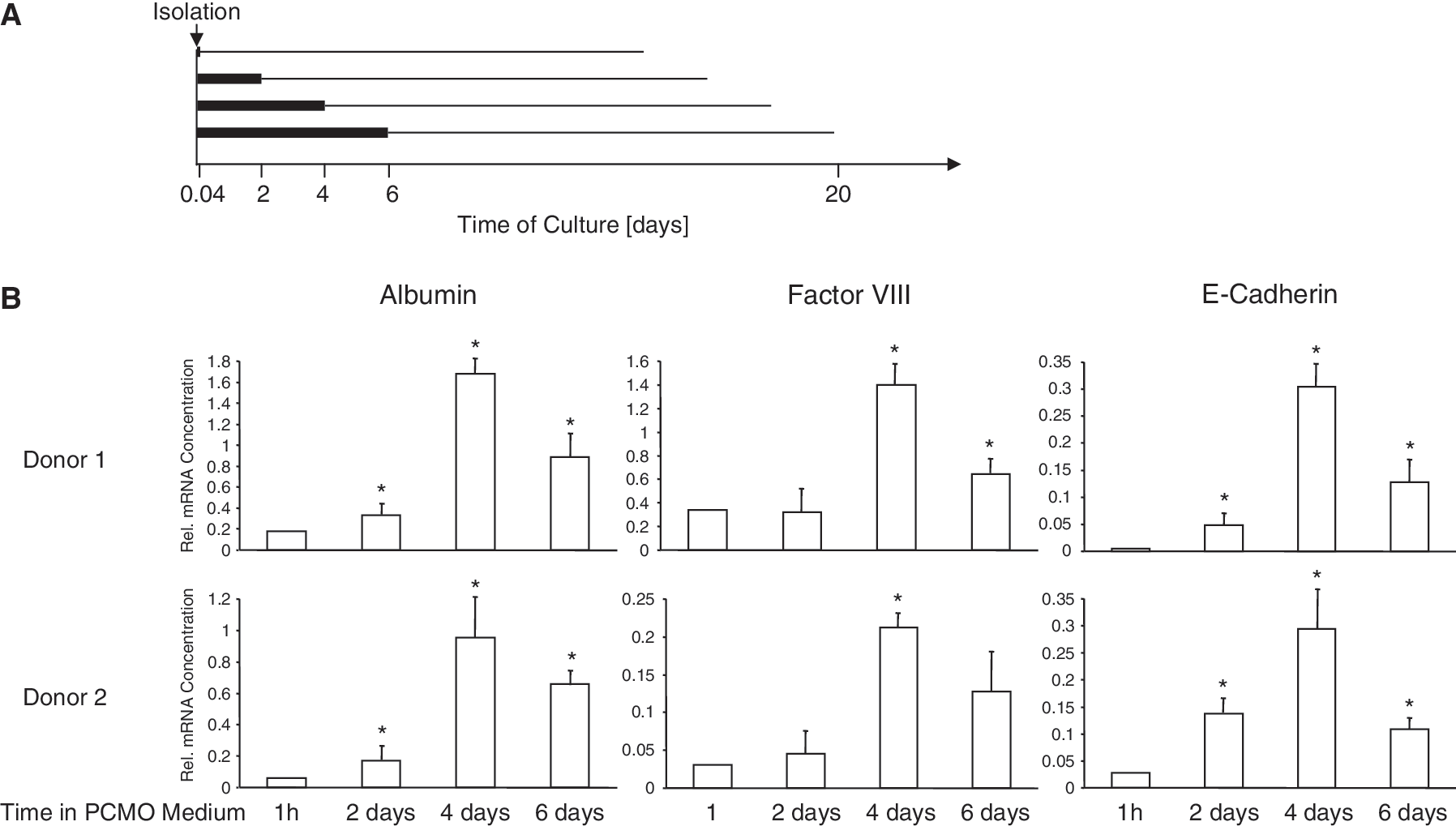
Optimal hepatocytic differentiation of programmable cells of monocytic origins (PCMOs) varies with time in culture. (
Dedifferentiation and hepatic redifferentiation also proceed in highly purified monocyte preparations
Monocytes isolated by FC still contain ∼30%–40% CD14− cells, primarily T- and B cells, after adhesion, and this number is further reduced to ∼10%–20% by Day 6 (Ref. [5] and unpublished data). In order to test whether the generation of PCMOs from CD14+ monocytes was dependent on the presence of these “contaminating” cells (acting on PCMOs through either cell–cell contacts or soluble factors), we subjected highly purified cultures (>95%) of CD14+ monocytes derived by elutriation to the same dedifferentiation–NeoHep differentiation protocol. We observed down-regulation of PRDMI (Fig. 3A) and ICSBP, but constant expression of the promyeloid Ets-family transcription factor Pu.1 (data not shown) as previously described in FC-purified preparations [5]. TLR expression, as exemplified here by TLR4, was rapidly down-regulated (Fig. 3A). Recently, Krüppel-like factor 4 (Klf4) was identified to be expressed in a monocyte-restricted and stage-specific pattern during myelopoiesis and to promote monocyte differentiation [18]. Klf4 was rapidly silenced following exposure of monocytes to PCMO culture conditions and transcript levels remained low until Day 6 (Fig. 3A) after which they became undetectable. The proliferation-associated c-MYC gene was transiently activated with peak expression on Day 2 (Fig. 3A). The reciprocal expression of PRDMI and MYC during the first 2 days of PCMO culture may indicate reactivation of MYC as consequence of its derepression from PRDMI, since ectopic Blimp-1, the murine homolog of human PRDMI, repressed myc expression and inhibited cell growth [19]. Expression of ID2, which is induced by serum and acts as an inhibitor of cellular differentiation [20], peaked on Day 2 and subsequently declined (Fig. 3A). Expression of KDR, a marker of endothelial progenitor cells, was generally low, but appeared to be regulated peaking at the 48-h time point (Fig. 3A). Transcript levels of CD105/endoglin, a marker of both ESCs and endothelial progenitors, were elevated between 24 and 72 h of culture and on Day 6 (Fig. 3A). In a second series of experiments, we successively cultured elutriated monocytes (5 different preparations) for up to 6 days in PCMO medium prior to NeoHep differentiation and subsequently determined albumin expression in these cells. One major “hotspot” of differentiation sensitivity was noted early on Day 2 of culture (Fig. 3B). A similar profile was found for the Factor VIII gene (data not shown). These data indicate that monocyte dedifferentiation is independent from the presence of CD14− cells and that under these conditions the cells more rapidly acquire a plastic state.
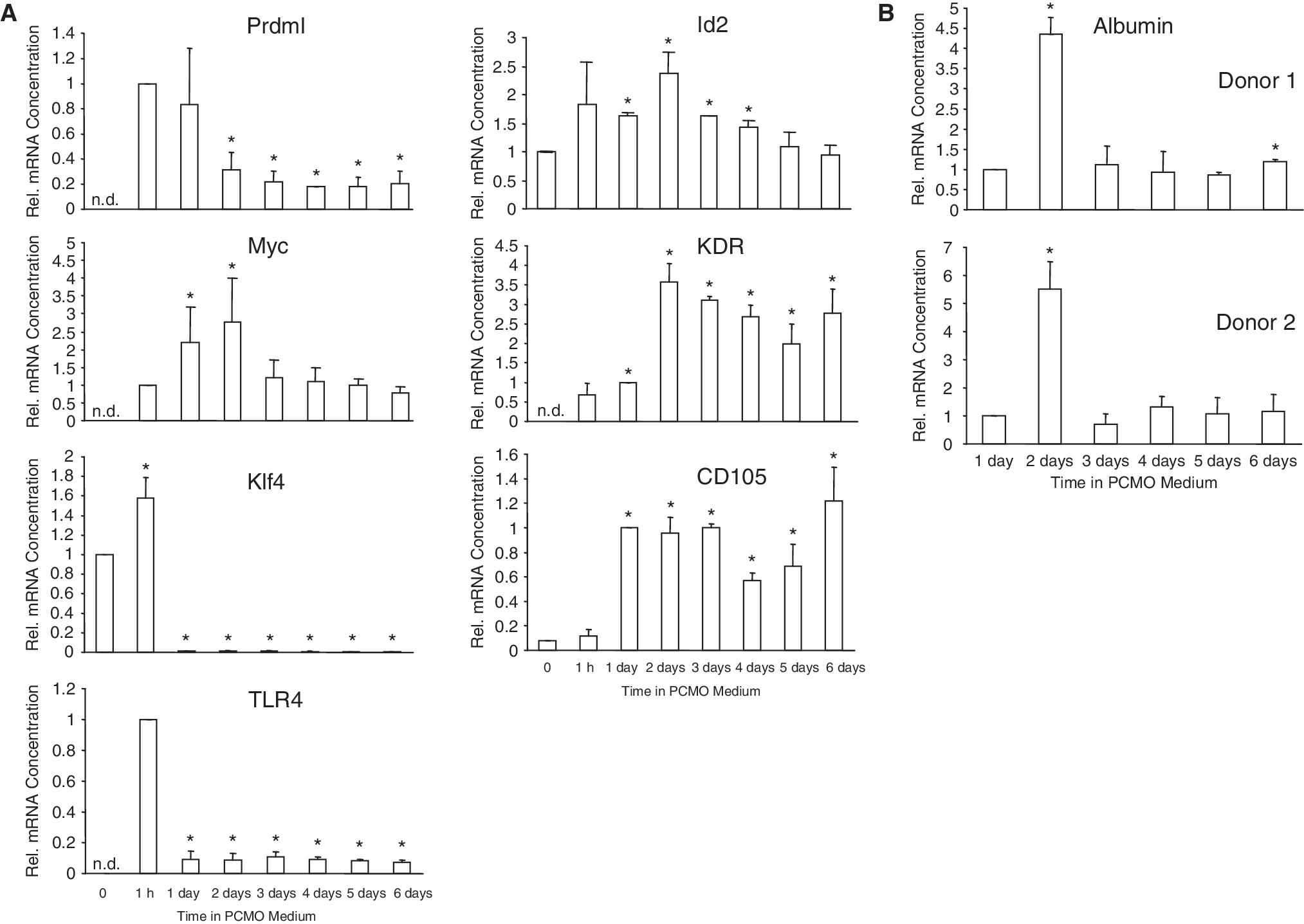
Dedifferentiation and hepatic redifferentiation proceeds in highly purified monocyte preparations. (
PCMOs express hES markers
The time-dependent pattern of sensitivity toward hepatocytic differentiation likely reflects underlying changes in multipotency and may be regulated by genes known to control self-renewal and pluripotency in ESCs. RT-PCR-based screening of FC-purified and elutriated monocytes revealed that on Day 6 of culture cells were consistently positive for OCT4, REX1, and NANOG, while SOX2 was negative in all (5/5) preparations tested (Fig. 4A). The failure to detect SOX2 confirmed the absence of other stem cell types such as HSCs, mesenchymal stem cells (MSCs), and neural stem cells in the PCMO cultures. Due to the lack of introns in OCT4 and NANOG and the presence of pseudogenes in the human genome, we have carefully evaluated that the signal was derived from mRNA rather than genomic DNA contamination and by using primers that do not recognize pseudogenes [13]. OCT4 expression was also verified at the protein level following immunoprecipitation and subsequent immunoblotting of total cellular proteins with a monoclonal antibody recognizing a single epitope localized at amino acids 1–134 (Fig. 4B), thus precluding detection of the OCT4B isoform ([21] and see below). Prior enrichment of nuclear and cytoplasmic proteins from PCMOs followed by direct immunoblotting revealed that both OCT4A and NANOG proteins were more abundant in the nuclear fraction (Fig. 4C).
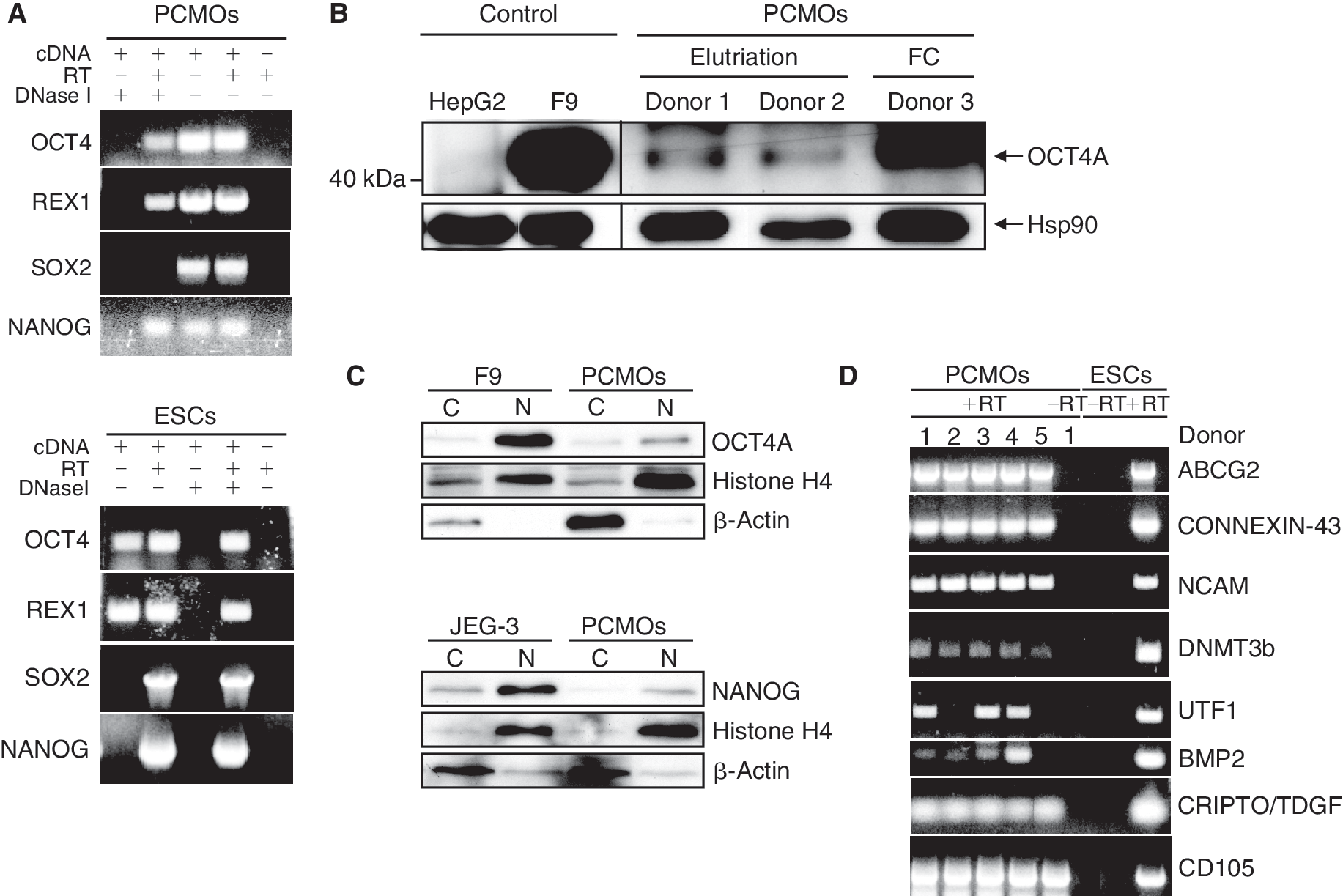
Expression of pluripotency markers in programmable cells of monocytic origins (PCMOs). (
Other genes besides c-MYC and KLF4 implicated in maintenance of self-renewal and pluripotency and to cluster with hESCs [22] and induced pluripotent stem (iPS) cells were expressed in PCMOs generated from elutriated monocytes and include (positive/total number of donors analyzed): reduced expression 1 (REX1) (3/5), growth and differentiation factor 3 GDF3 (3/5), DPPA3/STELLA/PGC7 (4/5) (not shown), ABCG2 (5/5), Connexin-43 (5/5), NCAM (5/5), DNMT3b (5/5), UTF1 (3/5), BMP2 (4/5), CRIPTO/TDGF1 (5/5), E-CADHERIN (5/5), and CD105 (5/5) (Fig. 4D). QPCR analysis showed that these genes were expressed at a much lower level than in NeoHeps, while others like GABRB3, NODAL, LEFTY B, and telomerase reverse transcriptase (hTERT) were negative in both PCMOs and NeoHeps from all 5 preparations analyzed (data not shown).
Reactivation of pluripotency markers and global histone H3 Lys-4 methylation during PCMO generation
Two different mRNAs are generated from the OCT4 gene by alternative splicing and encode 2 isoforms designated OCT4A and OCT4B [23]. Only OCT4A is associated with pluripotency, but not the related OCT4B, which lacks sequences encoded by exon 1 [21,24]. To distinguish between both isoforms, we monitored OCT4 expression at various time points during the monocyte → PCMO transition in elutriated monocytes using 2 different upstream primers. One recognized both transcripts (OCT4A + OCTB), while the other one bound to exon 1 and specifically amplified OCT4A. We observed that expression of both total OCT4 and OCT4A peaked on Days 2–3 and subsequently declined (Fig. 5A), and was undetectable in NeoHeps (data not shown). Notably, OCT4A expression correlated temporally with the susceptibility “hotspot” to hepatic differentiation (compare Fig. 3B). Interestingly, NANOG expression in elutriated monocytes followed a similar time course, but was slightly delayed relative to that of OCT4 (Fig. 5B). The data indicate that critical pluripotency regulators (eg, OCT4, NANOG) and possibly other factors of the pluripotency network are reactivated in PCMOs and are likely to control their plastic behavior.

Kinetics of OCT4 and NANOG activation and histone H3(K4) methylation in programmable cells of monocytic origins (PCMOs). (
Histone methylation plays a crucial role in epigenetic regulation of gene expression during mammalian development. In general, transcribed genes are associated with trimethylation at Lys-4 of histone H3 (me-H3(K4)) [25], whereas many silenced genes are associated with H3(K27) trimethylation [26]. In undifferentiated cells, the OCT4 promoter is packaged with nucleosomes that contain markers of active chromatin, namely histone H3 highly acetylated at Lys-9 and Lys-14, and me-H3(K4) [27,28]. In both adherent FC-purified monocytes (Fig. 5C) and in PCMO cultures from elutriated monocytes (data not shown), we noted a transient rise in global me-H3(K4) that closely mirrored the time course of OCT4 transcriptional activity. The methylation of the OCT4 promoter remained high during various stages of PCMO generation, as revealed by bisulfite conversion and pyrosequencing at specific CpG islands in the OCT4 distal enhancer [29] (Supplementary Fig. 1; Supplementary materials are available online at
Discussion
In this study, we have analyzed in more detail the molecular changes that occurred during the conversion of freshly isolated peripheral blood monocytes to PCMOs. We noted rapid down-regulation of TLRs 2, 4, 7, and 9, NOX4, p47phox, and KLF4, suggesting reduced capacity for microbial sensing/killing, and reduced ability for proinflammatory signaling and differentiation in these cells [30,31]. The role of KLF4 is particularly interesting, since its forced expression in primary CMPs or HSCs induced exclusive monocyte differentiation and activated the CD14 promoter [18]. Hence, its down-regulation in PCMOs may be a prerequisite for dedifferentiation and may account for the partial loss of CD14 expression (see Fig. 1A). However, since KLF4 is also essential for efficient genetic reprogramming in the process of induced pluripotency [32], residual KLF4 along with OCT4 [32] is likely required for PCMOs to acquire a multipotent state. Interestingly, however (ectopic) expression of OCT4 in certain somatic cells has also been associated with active dedifferentiation [33]. The expression patterns of the above-mentioned differentiation markers and of the ID2 and MYC genes (see Fig. 3A) strengthen our hypothesis that the culture conditions applied induce monocytes to acquire a more undifferentiated phenotype.
Studies with ex vivo Flt3 ligand-expanded murine monocyte/macrophage precursors showed that their intrinsic sensitivity toward exogenous in vitro differentiation to a specific phenotype changes with time in culture [17]. This mirrors a sequential commitment to macrophage (M-CSF), osteoclast (M-CSF + RANKL), dendritic cell (GM-CSF + TNF-α), or microglia (glial cell-conditioned medium) differentiation in response to the respective differentiation agents, while the differentiation potential to macrophages was constitutive [17]. This suggests that the macrophage phenotype is the default pathway and that windows for optimal differentiation toward other phenotypes also exist in cultured human monocytes. Indeed, results of this study showed that within the time frame analyzed monocytes exhibited a peak of increased sensitivity to external (hepatocytic) differentiation cues; peak plasticity was recorded on Day 4 in FC-purified monocytes and on Day 2 in elutriated monocytes. Since in FC-derived monocytes the 4-day time point correlated with maximal mitotic activity [5], it is tempting to speculate that proliferation is a prerequisite for the cells to adopt a nonhematopoietic phenotype. Dooner and coworkers indeed demonstrated that marrow cells with the capacity to convert into cells with a pulmonary epithelial phenotype after transplantation showed an increase with cytokine (eg, IL-3)-induced cell cycle transit [34].
As discussed earlier, our culture conditions apparently allowed for limited self-renewal capacity and considerable dedifferentiation of monocytes toward a more primitive progenitor that shares some features with the progenitor of monocytes and granulocytes (GMP) [5]. Interestingly, Charrière and coworkers found similarities between transcriptomic profiles of macrophages and murine undifferentiated ESCs, but not differentiated stem cells [35]. Dedifferentiation combined with proliferation is also the immediate response toward induction of the reprogramming factors OCT4, SOX2, KLF4, and c-MYC in the process of “induced pluripotency” in terminally differentiated cells [36]. This prompted us to hypothesize that PCMO generation involved genetic reprogramming events including reactivation of ESC markers and/or genes regulating pluripotency and self-renewal. In elutriated monocyte preparations we have detected, amongst others, mRNA encoding OCT4, KLF4, MYC, NANOG, and the OCT4 target genes REX1 and UTF1, the notable exception being SOX2. Intriguingly, hotspots of differentiation sensitivity indeed correlated not only with elevated expression of MYC and ID2 (markers of proliferation and suppressed differentiation, respectively), but also with transiently induced OCT4 and NANOG in the cultures (compare Figs. 3 and 5). So far, OCT4 expression in other multipotential subsets of monocytes has only been reported for monocytic endothelial progenitor cells [13] and in PBMCs at both the mRNA and protein levels [37]. However, the tools used by these authors preclude definite conclusions as to whether the A isoform of OCT4 (the only isoform associated with pluripotency [21,24]) has been detected. By using PCR primers for exon 1 and a monoclonal antibody recognizing the exon 1-encoded N-terminal epitope, we were able to demonstrate transcriptional induction of OCT4A during exposure of CD14+ cells to M-CSF, IL-3 and serum. The finding that elevated OCT4(A) expression coincided with both the onset of proliferation, up-regulation of the proliferation-associated MYC gene, and the time of peak sensitivity to external hepatocyte differentiation clues clearly argues in favor of OCT4 determining multipotency in PCMOs and we are currently performing functional studies to test this assumption. However, in light of recent doubts on such a role in adult somatic cells [38], alternative explanations have to be considered. One possibility is that OCT4 has a role in driving cell cycle progression (as reported for MSCs [39]) rather than in conferring pluripotency, and that its up-regulation in cycling cells or during specific phases of the cell cycle allows for a higher sensitivity to external differentiation clues merely as a “collateral effect.” It may also be possible that the reactivation of OCT4 in monocytes/PCMOs in response to culture conditions does not reflect a physiological mechanism in place to maintain somatic stem cells in a undifferentiated state in vivo, but may nevertheless result in a therapeutically relevant cell type [38].
PCMOs appear to display several features of partially reprogrammed cell lines [36] such as partial reactivation of genes related to stem cell renewal and maintenance (such as MYC), and some pluripotency genes (OCT4, NANOG), and incomplete repression of lineage-specifying transcription factors (such as PU.1 [5]). A third feature of stable partially reprogrammed cell lines is DNA hypermethylation at pluripotency-related loci. Interestingly, the distal enhancer region of OCT4 remained highly DNA-methylated in PCMOs (see Supplementary Fig. 1). Currently, it is not known for PCMOs whether histone H3 Lys-4 is methylated and H3 Lys-27 is demethylated in the promoter regions of OCT4 and NANOG as in human iPS cells [40]. Based on these observations it may be possible to further enhance genetic reprogramming of PCMOs and eventually induce a pluripotent state through (1) genetic complementation of SOX2, (2) silencing of PU.1, which has recently been shown to repress stem cell capacities in HSCs [41], or DNMT1, or (3) treatment of PCMOs with small-molecule compounds that modify chromatin or enhance the action of pluripotency factors [42].
It is currently unclear whether the fluctuation(s) in OCT4 and NANOG expression reflect transcriptional regulation of these genes in the majority of cells in a given culture, or whether it is the result of preferential expansion of a subpopulation of CD14+ cells with constitutively high expression of these markers. The dramatic changes seen in histone modifications during PCMO generation clearly argue in favor of the first scenario. An assembly of available data on cultured human cell populations that originate from circulating monocytes and have the potential to differentiate into nonphagocytes [43] has shown that these monocyte derivatives share the expression of CD14, CD45, and CD34. With respect to expression of CD34 detected by conventional flow cytometry (on a minor fraction [5]) and qPCR, and KDR detected by qPCR (this study), PCMOs resemble the CD14+CD34lowKDR+ subset described by Romagnani and colleagues [13]. PCMOs, like pluripotent stem cells (PSCs) [44], require M-CSF, but unlike monocyte-derived multipotential cells (MOMCs) [45], they require neither fibronectin (H. U., unpublished data) nor factors secreted by CD14− cells. A transient pattern of PCMO sensitivity to NeoHep differentiation was observed in both standard FC-purified monocyte cultures (containing a variable number of CD14− cells) and highly pure preparations of CD14+ cells derived by elutriation. However, peak sensitivity in crude cultures was delayed by ∼48 h, suggesting that CD14− cells in monocyte/PCMO cultures inhibit the rapid acquisition of a multipotent state. Activated T cells, for instance, may interfere with dedifferentiation of monocytes by promoting their differentiation toward osteoclasts via the expression of RANKL [46].
Results from the present study and a previous publication [5] show that monocyte-derived NeoHeps are generated in a 2-step process involving dedifferentiation with passage of the monocyte through a stem cell-like progenitor, and subsequent redifferentiation. We therefore favor dedifferentiation rather than transdifferentiation as the underlying mechanism, given that both mechanisms have been recognized as separate entities [10]. Dedifferentiation of cells in adult mammals with subsequent crossing of lineage boundaries has not been clearly and unequivocally documented [10]; however, there is increasing evidence that under physiologic conditions cell dedifferentiation exists in reversible equilibrium with differentiation. For instance, in hematopoietic development postnatal hemangioblasts are generated by dedifferentiation of committed HSCs [47]. We envision that both phenotype and metabolic function of NeoHeps, and possibly other tissue-specific PCMO derivatives, can be improved by optimizing the conditions for cell-type-specific differentiation, and, even more effectively, by enhancing the PCMOs' plasticity through the above-mentioned genetic and epigenetic strategies.
Footnotes
Acknowledgments
We are indebted to J. Richter, M.Sc. (Institute of Human Genetics, UKSH, Campus Kiel) for performing the Bisulfite-Pyrosequencing analysis of OCT4 and I. Berg, M. Jansen, and K. Kopp for excellent technical assistance. We thank Drs. P. Björquist and N. Heins (CellArtis, Stockholm, Sweden) for donating RNA of the human ESC line SA002.5. This work was supported in part by a grant from the “Bundesministerium für Bildung und Forschung” (01 GN 0985).
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.