
Abstract
Mesenchymal stem cells (MSCs) inhibit the proliferation of allo-activated lymphocytes. This effect is primarily dependent on the secretion of anti-inflammatory factors by MSCs and is enhanced under inflammatory conditions. MSCs, however, also produce factors that can potentially activate resting immune cells. Full understanding of the behavior of MSCs under inflammatory and noninflammatory conditions is crucial when clinical application of MSCs is considered. Human adipose tissue-derived MSCs were cultured with nonactivated peripheral blood mononuclear cells (PBMCs) and the activation, proliferation, and function of PBMCs were examined. Seven days of coculture with autologous or allogeneic MSCs significantly increased the proliferation of PBMCs (3-fold). This effect was observed in both direct and transwell coculture systems. MSCs cocultured with PBMCs showed increased mRNA expression of the proinflammatory mediators interleukin-6 (IL-6), IL-8, tumor necrosis factor-α, the growth factors basic fibroblast growth factor and vascular endothelial growth factor-α, and the anti-inflammatory factor indoleamine 2,3-dioxygenase. After removal of MSCs, PBMCs showed a spectacular further increase in proliferation, with a maximum of 25-fold after 7 days. This increase in proliferation was not seen when PBMCs were kept in the presence of MSCs. The proliferating fraction of PBMCs largely consisted of CD4+ T-cells with high CD25 expression and the proportion of CD127negFoxP3+ regulatory T-cells significantly increased from 5.0% to 8.5% of total CD4+ T-cells. The expanded T-cells demonstrated normal responses to mitogen or alloantigen stimulation. The CD25positive fraction of these cells had immunosuppressive capacity. In conclusion, MSCs can stimulate the activation and proliferation of resting T-cells and generate regulatory T-cells. These findings are important when MSCs are applied in the clinic.
Introduction
O
Animal models have demonstrated that MSCs can effectively reduce inflammatory processes, for example, in autoimmune encephalitis [8], myocarditis [9], inflammatory bowel disease [10], and glomerulonephritis [11]. In a clinical setting, MSC therapy has been shown to be effective for the treatment of patients with severe therapy-resistant graft-versus-host disease [12]. Further, clinical trials are ongoing to determine whether MSCs are also effective for the treatment of autoimmune diseases, such as inflammatory bowel diseases, multiple sclerosis, and rheumatoid arthritis.
The immunosuppressive properties of MSCs make them interesting candidates for cell-based therapy in organ transplantation [13]. Treatment with MSCs may provide an immunosuppressive environment that controls antidonor reactivity. A number of studies with animal transplant models have reported a beneficial effect of MSCs on skin [14], heart [6,15], and liver [16] graft survival.
We recently reported that MSCs derived from perirenal adipose tissue of living kidney donors have the capacity to inhibit antidonor reactivity of kidney transplant patients in vitro, both before and after transplantation [17]. The immunosuppressive function of MSCs is triggered under inflammatory conditions where T-cells produce interferon-γ (IFNγ) and tumor necrosis factor-α (TNFα) [18,19]. The survival of T-cells in a quiescent state, however, is promoted by MSCs [20]. Despite many studies investigating the effect of MSCs on alloantigen or mitogen-activated immune cells, the effect of MSCs on nonactivated immune cells remains largely unknown. Understanding the full spectrum of immune modulation by MSCs is, however, important when the application of MSCs in a clinical setting is considered. In the present study, we investigate the effect of human autologous and allogeneic MSCs on nonactivated immune cells.
Materials and Methods
Isolation, culture, and differentiation of MSCs from perirenal adipose tissue
MSCs were isolated from 10 to 50 g of perirenal adipose tissue of living kidney donors as previously described [21]. Briefly, perirenal adipose tissue was collected during the kidney donation procedure after written informed consent of the kidney donors as approved by the Medical Ethical Committee of the Erasmus Medical Center Rotterdam (protocol no. MEC-2006-190). The tissue was collected in RPMI-Dutch-modified-1640 (RPMI; Invitrogen) with 200 μM
To confirm whether the perirenal adipose tissue-derived cells were indeed MSCs, they were characterized by flow cytometry, differentiated in osteogenic, adipogenic, and myogenic lineages, and added to mixed lymphocyte reactions (MLRs) to test their immunosuppressive capacity as previously described [17,21].
Isolation of peripheral blood mononuclear cells
Peripheral blood samples were collected from living kidney donors and from buffy coats of healthy blood donors. Peripheral blood mononuclear cells (PBMCs) were isolated from the collected heparinized peripheral blood by density gradient centrifugation using Ficoll Isopaque (δ = 1.077; Amersham) and frozen at −150°C until use.
Experimental setup (days 0–7): coculturing PBMCs with MSCs
The experimental setup is shown in Fig. 1. At day 0, PBMCs were cultured for 7 days with MSCs in a 5:1 ratio in direct or transwell coculture systems. In transwell coculture systems, PBMCs were separated from MSCs by a 0.4-μm pore-sized membrane (Greiner Bio-one); MSCs were cultured in the bottom well, while PBMCs were kept in the inserts. In 6-well plates, 1 × 106 PBMCs were added to 0.20 × 106 MSCs, whereas in 96-well plates, 50,000 PBMCs were added to 10,000 MSCs. After 7 days, proliferation was determined after 16 h incorporation of 3H-thymidine (0.5 μCi/well; Amersham) using a β-plate reader (LKB). In 6 of 56 cases, PBMCs in the absence of MSCs already showed 3H-thymidine incorporation of 5,000 cpm or more (day 7) and these samples were excluded from further analysis as these cells could not be defined as “nonactivated.”

Experimental setup. PBMCs were cocultured with MSCs in direct or transwell coculture systems during the first week (week 1). On day 7, MSCs were removed from the coculture systems, and culture of PBMCs continued in the absence of MSCs for another 7 days (week 2). On experiment day 14, the functionality of the cells previously cultured with MSCs was investigated (week 3). MSCs, mesenchymal stem cells; PBMCs, peripheral blood mononuclear cells.
In addition, proliferation was measured after 7 days by flow cytometry after labeling of PBMCs on day 0 with PKH67 (Sigma-Aldrich), according to the manufacturer's instructions. Flow cytometric analysis was performed as described later. To investigate whether IL-6 and IL-8 were involved in the stimulatory effect of MSCs on T-cell activation and proliferation, neutralizing antibodies against IL-6 (10 μg/mL) and IL-8 (10 μg/mL) (both R&D Systems) were used. Immunoassays (Immulite 1000; Siemens) were performed to determine the protein levels of IL-6 and IL-8 in the supernatants of cultures with neutralizing antibodies.
Experimental setup (days 7–17): continued culture of PBMCs previously cultured with MSCs
After 7 days, PBMCs were collected from PBMC–MSC cocultures, washed, and resuspended in fresh medium (RPMI with p/s,
Experimental setup (days 14–21): functional analysis of PBMCs previously cultured with MSCs
On experimental day 14, PBMCs were collected for real-time room temperature (RT)–polymerase chain reaction (PCR) analysis (as described later). To examine whether these cells were still capable of responding to alloantigens, they were used as responder cells and stimulated with 1 μg/mL phytohemagglutinin (PHA; Murex Biotech Ltd.) for 3 days or with γ-irradiated (40 Gy) allogeneic PBMCs (added at a 1:1 ratio).
Further, to determine whether the precultured PBMCs had immunosuppressive capacity, they were added to MLRs at a 1:10 ratio. Finally, the PBMCs were sorted for CD25 by auto magnetic-activated cell sorting (MACS) (as described later). The suppressive capacity of both the CD25pos and the CD25neg fractions was examined by adding the cells to MLRs at a 1:5, 1:10, and 1:20 ratios.
Mixed lymphocyte reactions
Immunosuppressive capacity of MSCs and PBMCs previously cultured with MSCs was tested in MLRs. In MLRs, 5 × 104 responder PBMCs were stimulated by 5 × 104 γ-irradiated (40 Gy) allogeneic PBMCs in RPMI + 10% HI-FBS in round-bottomed 96-well plates (Nunc). On day 7, proliferation was measured following incorporation of 3H-thymidine (0.5 μCi/well) during 16 h of incubation using a β-plate reader. To determine the proliferation capacity of the PBMCs, 5 × 104 cells were stimulated with 1 μg/mL PHA for 3 days, and 3H-thymidine incorporation was measured. Only results of responder PBMCs with sufficient proliferation capacity (>10,000 cpm after stimulation with PHA) were included.
Flow cytometric analysis
PBMCs were washed twice with FACSFlow (BD Biosciences), stained with antibodies against CD3-AmCyan, CD4-PacificBlue, CD8-APC-Cy7, CD16/56-APC, CD19-PerCP, CD25-PE-Cy7, CD62L-APC, CD69-PE, CD127-FITC (all from BD Biosciences), CCR7-PE (R&D Systems), and FAS-L-APC (Biocarta) at room temperature, and protected from light for 30 min. After 2 washes with FACSFlow, flow cytometric analysis was performed using an 8-color FACSCANTO-II with FACSDIVA Software (BD Biosciences). FoxP3-APC staining was carried out as described previously [22].
Isolation of CD25pos cells by autoMACS
On experimental day 14, the CD25pos cells were isolated from PBMCs that were previously transwell cocultured with MSCs using anti-CD25 microbeads (10 μL/10 × 106 cells) (Miltenyi Biotech), followed by positive selection (POSSELD program) on the autoMACS (Miltenyi Biotech). To check the purity, both fractions were stained with CD3-FITC, CD4-PerCP, and CD25-PE epitope B (clone M-A251; BD Biosciences) and flow cytometric analysis was performed.
Real-time RT-PCR
PBMCs and MSCs for real-time RT-PCR were obtained from transwell coculture systems. Total RNA was isolated and cDNA synthesized as previously described [23]. Quantitative gene expression was determined using TaqMan Universal PCR Master Mix and assays-on-demand for IDO (Hs00158627.m1), IL-10 (Hs00174086.m1), TGFβ (Hs00171257.m1), TNFα (Hs99999043.m1), CD25 (Hs00166229.m1), FoxP3 (Hs00203958), IL-2 (Hs00174114.m1), IL-6 (Hs00174131), IFNγ (Hs00174143), and IL-8 (Hs00174114.m1) (all from Applied Biosystems) on a StepOnePlus (Applied Biosystems). Vascular endothelial growth factor-α (VEGFα) and basic fibroblast growth factor (bFGF) were determined by real-time RT-PCR using homemade primers (forward, 5′ GCA GAC CAA AGA AAG ATA GAG CAA G 3′ and reverse, 5′ CGC CTC GGC TTG TCA CAT 3′; and forward, 5′ GTT GAC GGG GTC CGG G 3′ and reverse, 5′ GAT AGA CAC AAC TCC TCT CTC TTC TGC 3′, respectively). Gene expression levels were normalized to the concentration of 18S per 500 ng RNA.
Statistical analysis
Data were analyzed using paired t-test or Wilcoxon signed-rank test depending on the distribution of the data as tested with Kolmogorov–Smirnov test for normality. Parametric data are expressed as means, whereas nonparametric data are expressed as medians. Statistical significance was defined as P < 0.05 (2-tailed).
Results
MSCs stimulate PBMC proliferation and activation
In contrast to our previous studies [17,21], in which MSCs were added to allo-activated PBMCs, the present study investigated the effect of human MSCs on nonactivated PBMCs. The experimental setup is shown in Fig. 1.
Seven-day coculture of PBMCs with allogeneic MSCs significantly increased PBMC proliferation by a factor of 4, compared with PBMCs cultured without MSCs (Fig. 2A). This increase in PBMC proliferation was independent of HLA, because autologous MSCs also induced a 3-fold increase in PBMC proliferation. There was no significant difference in the proliferative effect of autologous and allogeneic MSCs. When PBMCs were cocultured with γ-irradiated MSCs, a similar increase in the proliferation of PBMCs was observed.
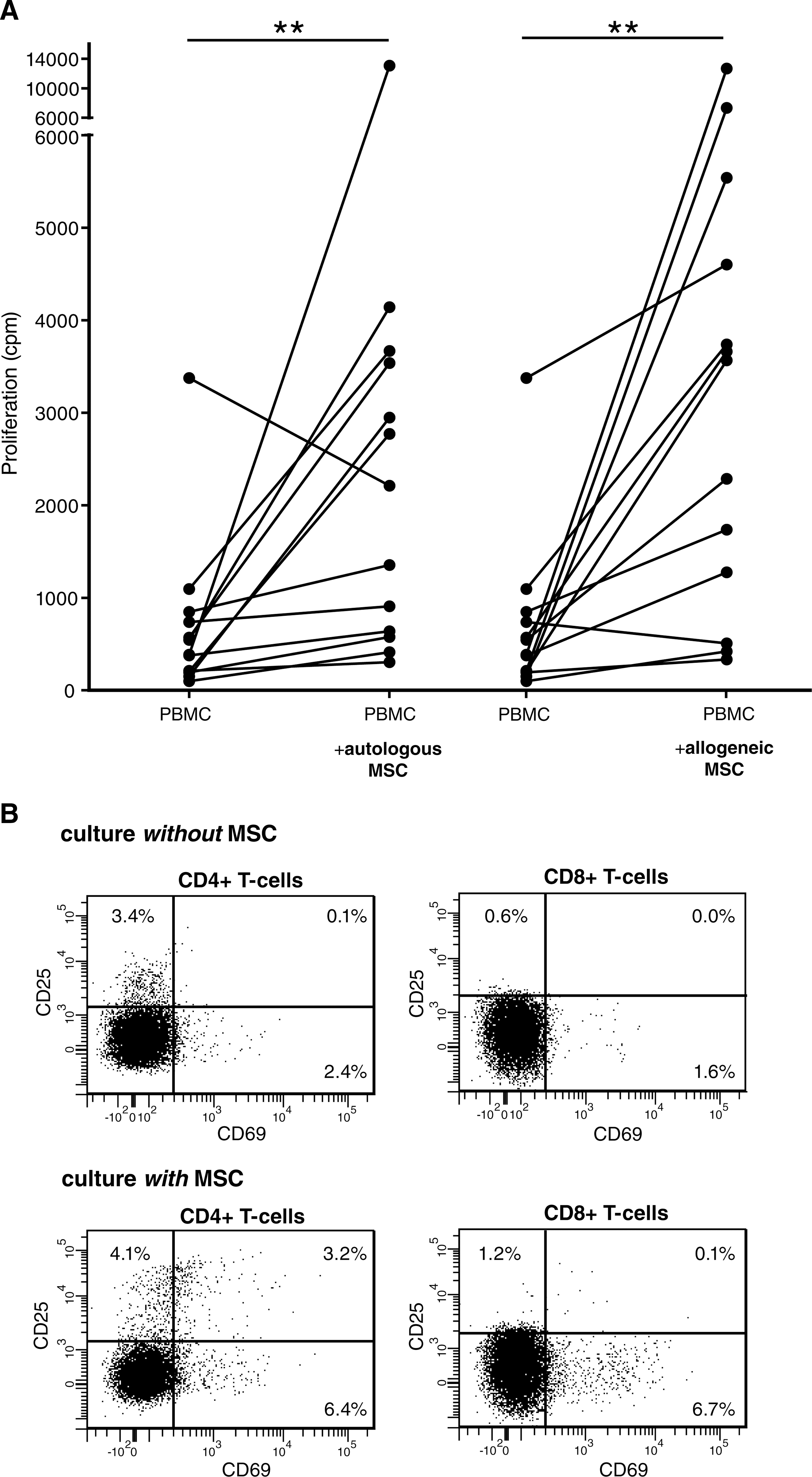
Proliferation and activation of PBMCs cultured for 7 days in the absence or presence of MSCs (in direct culture systems). (
To identify which PBMC subset's proliferation was stimulated in the presence of MSCs, flow cytometric analysis of PBMCs was carried out. MSCs predominantly stimulated the proliferation of CD4+ T-cells and CD8+ T-cells, whereas coculture with MSCs had no effect on the proliferation of natural killer (NK) and B-cells. Coculture of PBMCs with MSCs also significantly increased CD69 and CD25 expression on the proliferating CD4+ and CD8+ T-cells (Fig. 2B), whereas the expression of CD62L, FAS-L, and CCR7 was unchanged in T-cells after culture with MSCs. The increased CD25 expression of T-cells cocultured with MSCs was associated with a 2-fold increase in mRNA expression of IL-2 by the PBMCs (data not shown). The expression of proinflammatory cytokines such as IL-6, IL-8, IFNγ, or TNFα did not significantly change in PBMCs cocultured with MSCs. The expression of the immunosuppressive factor IL-10 decreased by a factor of 2 in PBMCs cocultured with MSCs (P = 0.0039).
The expression of proinflammatory, growth, and anti-inflammatory factors by MSCs is increased in the presence of PBMCs
The observed stimulatory effect of MSCs on PBMC proliferation was comparable in direct and transwell coculture systems, which indicates that the effect is mediated by soluble factors. To identify which factors may be responsible, gene expression of proinflammatory, growth, and immunosuppressive factors by MSCs was investigated (Fig. 3). MSCs significantly increased the expression of IL-8 (527-fold), IL-6 (11-fold), TNFα (3-fold), VEGFα (3-fold), and bFGF (4-fold) in the presence of PBMCs. In addition, the anti-inflammatory factor IDO was 699-fold increased in MSCs cultured with PBMCs. IL-2, IFNγ, and IL-10 were not detectable.
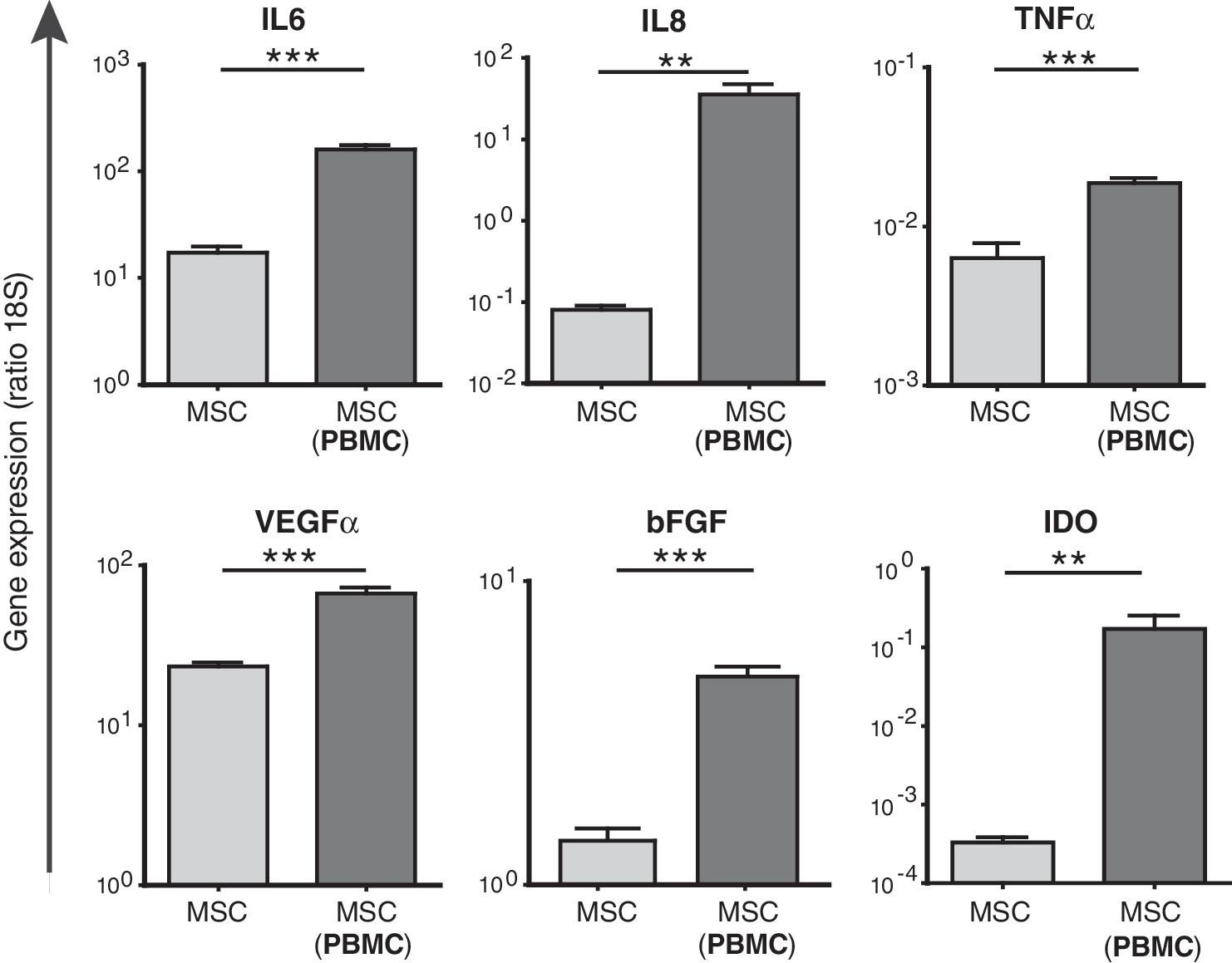
Real-time reverse-transcriptase (RT)-polymerase chain reaction analysis of MSCs cocultured with PBMCs for 7 days in transwell coculture systems. Gene expression levels were normalized to the concentration of 18S RNA per 500 ng RNA (n = 9). **P < 0.01; ***P < 0.001. MSCs, mesenchymal stem cells; PBMCs, peripheral blood mononuclear cells.
T-cell proliferation dramatically increases after removal of MSCs
To investigate whether the presence of MSCs was needed for continuation of PBMC proliferation, MSCs were removed from PBMC–MSC transwell coculture systems, and at the same time, PBMCs were continued in culture (Fig. 1, week 2). PBMC proliferation was measured at 1, 3, 5, 7, and 10 days after removal of MSCs. Continued culture of the PBMCs led to an explosive 25-fold increase in proliferation, with a maximum on day 7 after separation of PBMCs from MSCs (experiment day 14) (Fig. 4A). This extra increase in PBMC proliferation during the second week was not observed when PBMCs were kept in the presence of MSCs (Fig. 4B).
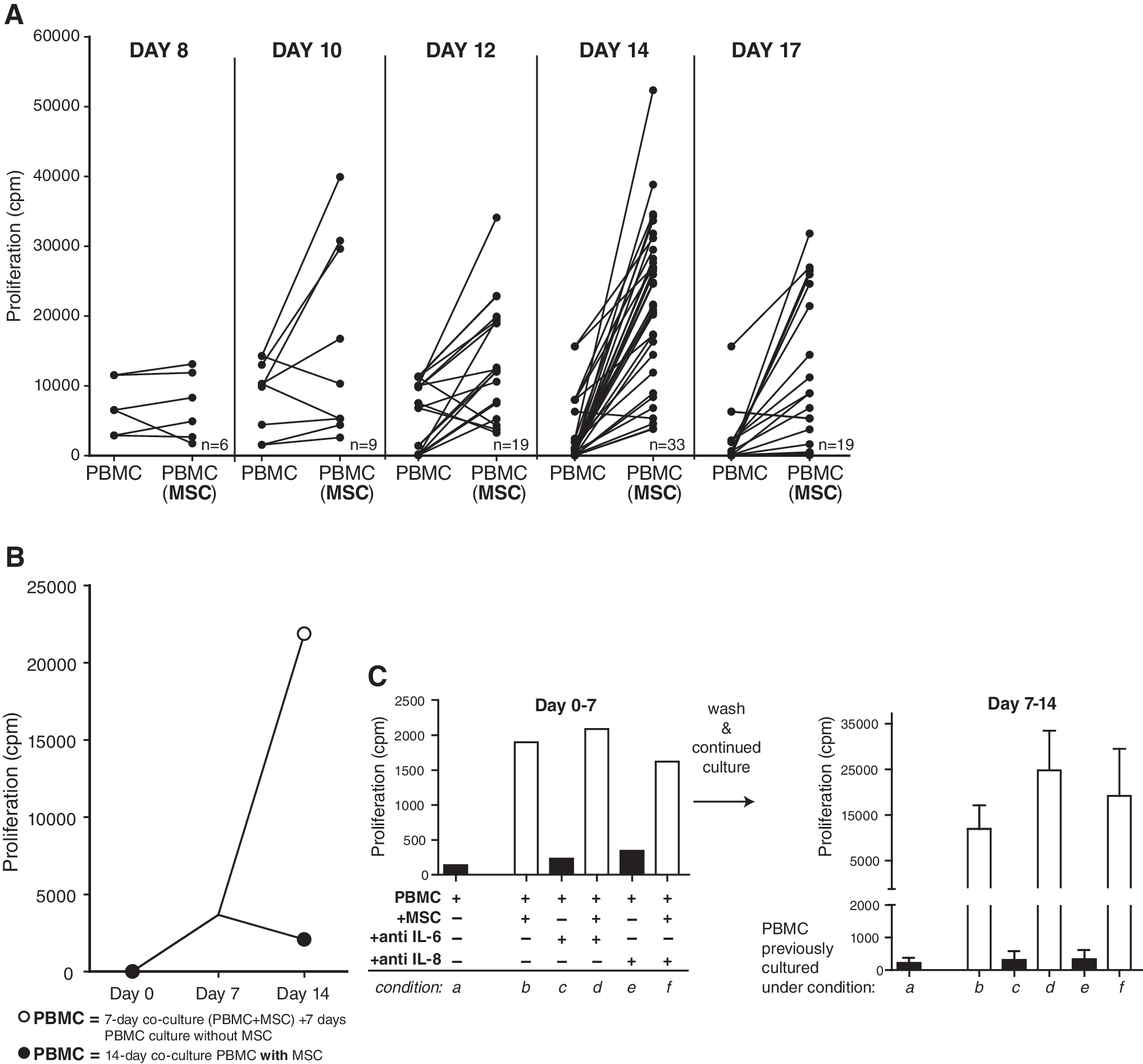
Continued culture of PBMCs previously transwell cultured with MSCs and after removal of MSCs at day 7. (

As shown, on experimental day 17, PBMC proliferation declined compared with day 14. To rule out the possibility that this was due to exhaustion of the culture medium, fresh medium was added on day 14 and PBMC proliferation was measured on day 17. Refreshment of medium, however, did not improve the proliferation of the PBMCs on day 17.
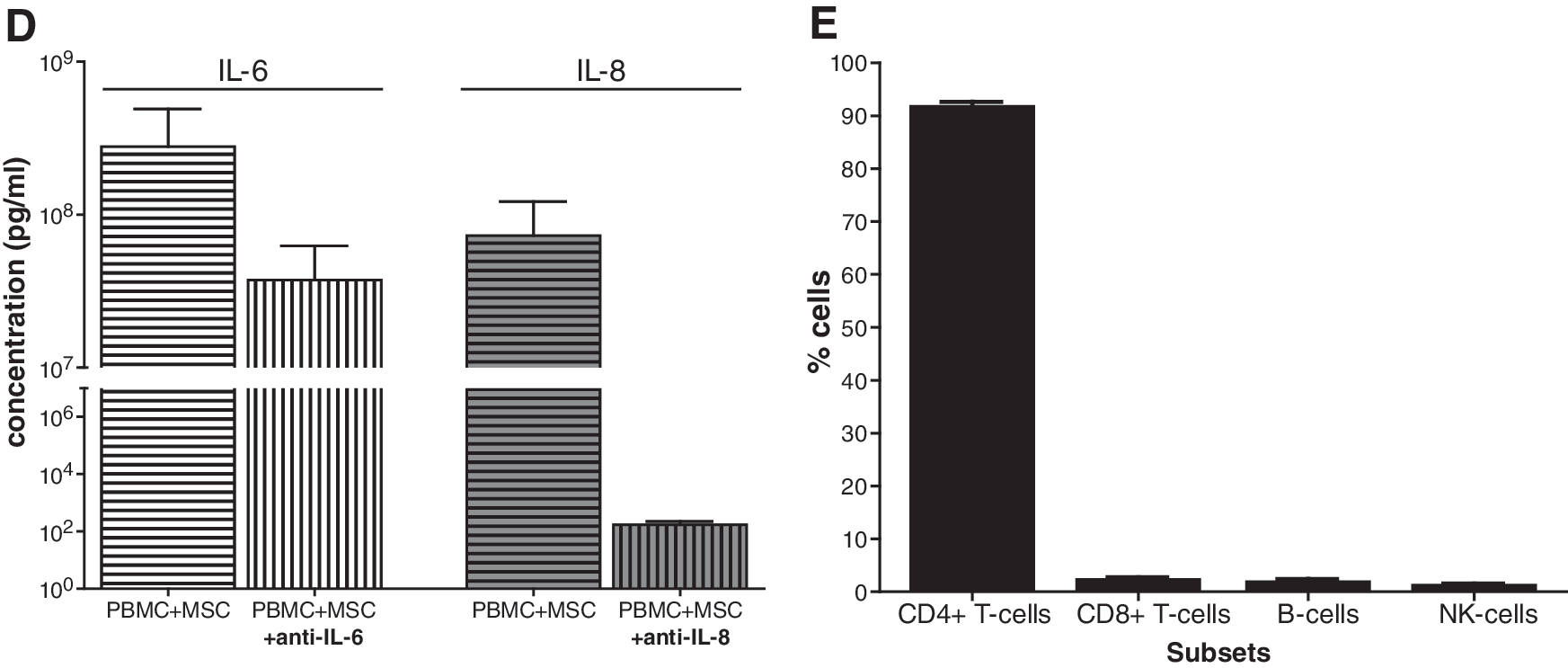
As the expression of IL-6 and especially IL-8 strongly increased in MSCs during culture with PBMCs, it was investigated whether IL-6 and IL-8 were involved in the stimulatory effect of MSCs on resting PBMCs. Therefore, PBMCs were cocultured with MSCs in the absence or presence of neutralizing antibodies against IL-6 or IL-8. Blocking IL-6 or IL-8 did not reverse the stimulatory effect of MSCs on PBMC proliferation (Fig. 4C). Protein levels of IL-6 and IL-8 in the supernatants were measured by immunoassays. Anti-IL-8 antibodies strongly reduced the IL-8 concentration in PBMC–MSC cocultures (4 × 105-fold). The production of IL-6 in PBMC–MSC cocultures was extremely high (3 × 108 pg/mL). Although anti-IL-6 antibodies reduced the IL-6 levels 7-fold, they were unable to completely remove free IL-6 (Fig. 4D).
MSCs induce activation and proliferation of CD4+ T-cells
Flow cytometric analysis was performed to identify which PBMC subset(s) exhibited the striking increase in proliferation observed at 7 days after removal of MSCs. The proliferating fraction of PBMCs expanded by MSCs consisted predominantly of CD4+ T-cells (92%) (Fig. 4E). The percentages of CD8+ T-cells, NK cells, and B-cells did not significantly change. The CD4+ T-cells previously cultured with MSCs showed a strong increase in proliferation (as measured by PKH dilution) (Fig. 4F). These cells highly expressed CD25 and CD69, whereas the expression of FAS-L, CD62L, and CCR7 was unchanged. Further, the percentages of CD4+CD25posFoxP3+ and CD127neg were significantly increased by a factor of 1.7, compared with PBMCs previously cultured without MSCs. This indicates that there is expansion of CD4+CD25posFoxP3+CD127neg Tregs in PBMCs previously cultured with MSCs.
Expression of pro- and anti-inflammatory genes in PBMCs after removal of MSCs
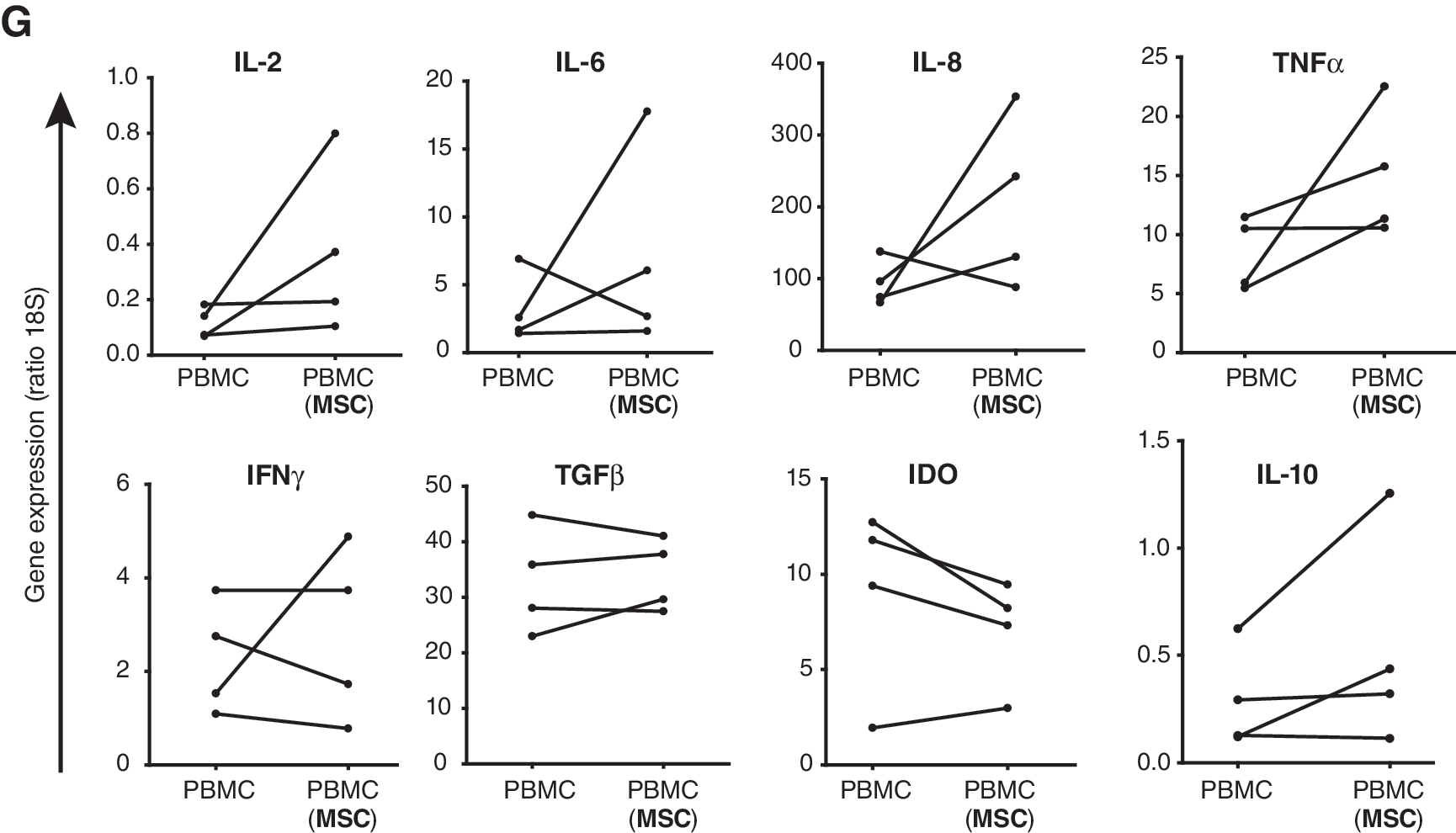
Next, PBMCs were collected at 7 days after removal of MSCs and RT-PCR analysis was performed to characterize the PBMCs expanded by MSCs (Fig. 4G). PBMCs expanded by MSCs showed significant, but minor, increases in the expression of proinflammatory factors IL-2 (3-fold), IL-6 (2-fold), IL-8 (2-fold), and TNFα (2-fold) compared with PBMCs not precultured with MSCs. There was no significant change in the expression of IFNγ or the anti-inflammatory factors TGFβ and IDO, whereas IL-10 expression increased by a factor of 2.
PBMCs expanded by MSCs remain responsive to mitogen and alloantigen
After 1 week of culture with MSCs, followed by 1 week of culture without MSCs, PBMCs were highly proliferative and showed an activated phenotype. To functionally examine the characteristics of these expanded cells, they were stimulated with mitogen (PHA) and alloantigen (γ-irradiated allogeneic PBMCs) (Fig. 1, week 3). PBMCs precultured with MSCs showed a good response upon stimulation with PHA (up to 42,000 cpm) (P = 0.005) and was comparable with the proliferative response of PBMCs cultured without MSCs. Thus, despite 2 weeks of culture, the expanded cells retained a good proliferative capacity.
PBMCs expanded by MSCs were then stimulated with γ-irradiated allogeneic PBMCs (at a 1:1 ratio). These PBMCs showed comparable proliferative responses compared with PBMCs previously cultured without MSCs. Thus, although the PBMCs previously cocultured with MSCs were highly activated and proliferative, the cells were not hyperreactive upon stimulation with alloantigen.
PBMCs expanded by MSCs have immunosuppressive capacity
We then investigated whether the expanded PBMCs precultured with MSCs had immunosuppressive capacity. Therefore, the PBMCs were added at a 1:10 ratio to MLRs (Fig. 5A). PBMCs that had been cultured with MSCs inhibited the proliferation in MLRs by 32% (P < 0.0001), whereas PBMCs previously cultured without MSCs did not show this effect.

Functionality of PBMCs previously transwell cultured with MSCs and continued in culture for another 7 days without MSCs. (
The inhibitory capacity of the PBMCs was independent of the origin of the cells used in the MLRs, indicating that inhibition was not antigen specific. PBMCs that were cultured for 1 week with MSCs and added to MLRs before their expansion had no immunosuppressive capacity, indicating that the expansion of these cells is crucial for obtaining their immunosuppressive capacity.
The immunosuppressive capacity of PBMCs expanded by MSCs resides in the CD25pos T-cell fraction
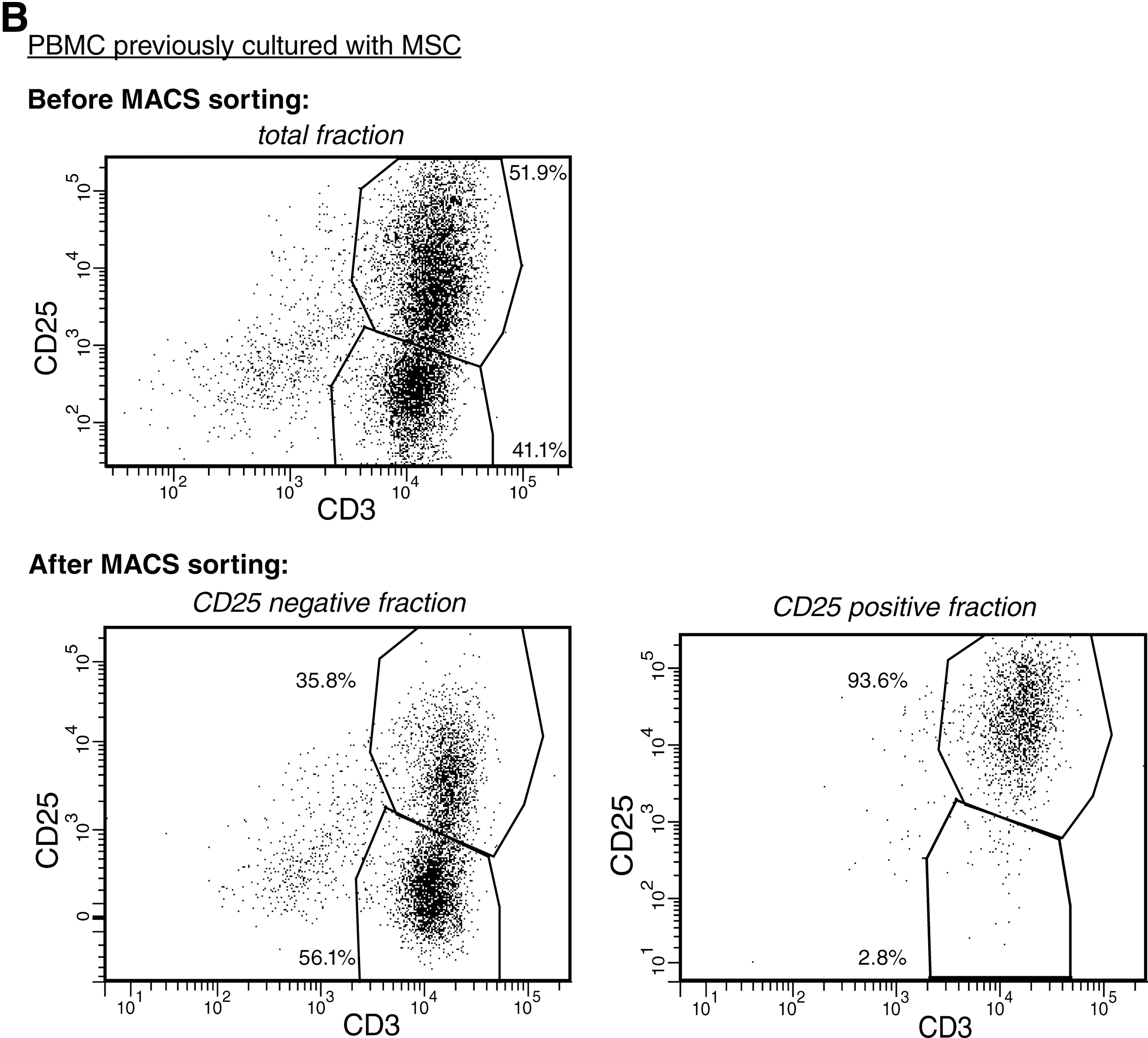
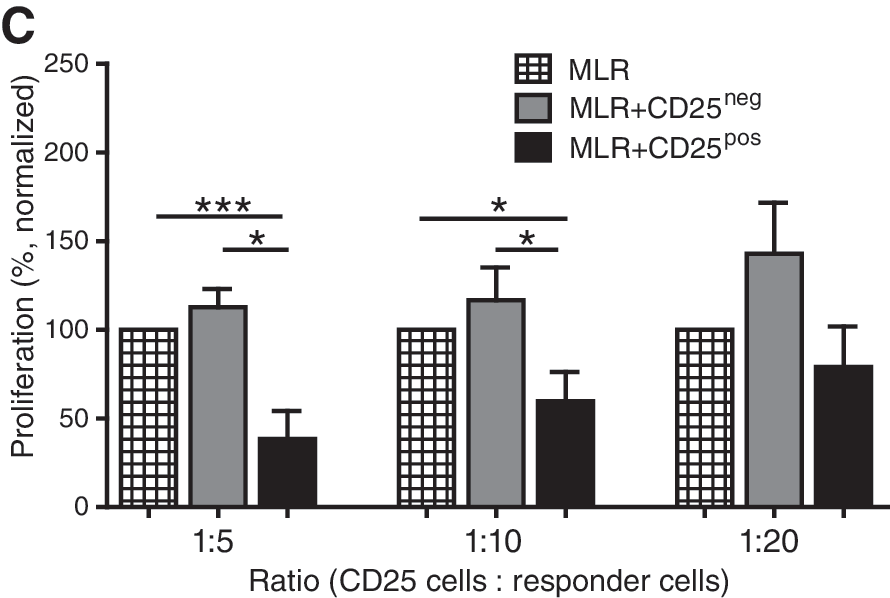
Finally, the previous experiments showed that MSCs induced both CD25 expression and immunosuppressive function in PBMCs. To examine whether the CD25pos PBMCs were responsible for the observed immunosuppressive capacity, PBMCs expanded by MSCs were sorted in CD25pos and CD25neg fractions and added to MLRs at various ratios. Before sorting, 51.9% of the PBMCs previously cultured with MSCs were positive for CD25. After sorting, the purity of CD25pos cells was 93.6% (Fig. 5B). The CD25neg fraction had no inhibitory capacity at any ratio and even stimulated the proliferation of allo-activated PBMCs, whereas the CD25pos fraction significantly inhibited the proliferation of allo-activated responder cells in MLRs by 62% (ratio 1:5), 40% (ratio 1:10), and 21% (ratio 1:20) (Fig. 5C).
Discussion
There is accumulating data on the immunosuppressive effect of MSCs demonstrating that MSCs inhibit mitogen and alloantigen-activated immune cells. The effect of MSCs on immune cells under low immunogenic conditions is, however, unknown. It is important to understand the full spectrum of immune regulation by MSCs, when MSCs are considered for clinical application.
The present study demonstrates that the proliferation of nonactivated, “resting” PBMCs increases by coculture with MSCs. Removal of MSCs leads to accelerated proliferation of, in particular, CD4+ T-cells, whereas in the continuous presence of MSCs, PBMC proliferation is kept at a low level. This suggests that MSCs activate resting CD4+ T-cells, but, when present, do prevent full activation and proliferation of these cells.
These results are in sharp contrast with previous studies reporting that MSCs do not elicit proliferative responses from allogeneic lymphocytes [24 –27]. One of the reasons for this disparity is that most groups investigated the effect of MSCs on activated PBMCs, whereas the present study is the first to investigate the effect of MSCs on nonactivated PBMCs in detail. Further, this is the first study to examine the effect of removal of MSCs from MSC–PBMC cocultures. It can be speculated that the observed stimulatory effect of MSCs on resting T-cells is still present in an activated setting, but that it is covered by the immunosuppressive effect of MSCs on activated PBMCs.
It is generally considered that T-cell activation and proliferation is initiated by T-cell receptor triggering. In our study, however, we found that the activation was not antigen dependent, as the proliferation of T-cells was stimulated by both autologous and allogeneic MSCs, and that this effect was present in transwell systems, indicating that T-cells can be activated by soluble factors only. Apart from the repeatedly reported production of anti-inflammatory factors [1 –3], MSCs can also produce growth and proinflammatory factors, such as bFGF, IL-6, and TNFα, which can explain the stimulatory effect of MSCs on PBMCs [28]. In the present study, gene expression analysis showed that MSCs had particularly increased levels of IL-6 and IL-8 after coculture with PBMCs. Neutralizing IL-8 dramatically reduced the IL-8 concentration in the supernatants of PBMC–MSC cocultures but had no effect on PBMC proliferation, suggesting that IL-8 is not a major player involved in the stimulatory effect of MSCs on PBMCs.
In addition, reduction of IL-6 levels by neutralizing antibodies in the PBMC–MSC cocultures did not prevent the activation and proliferation of resting PBMCs. However, remaining levels of IL-6 could still be responsible for the stimulatory effect of MSCs on PBMCs. We can therefore not rule out that the stimulatory effect of MSCs on PBMCs was independent of IL-6 and this needs further investigation.
Concurrent with the increased expression of IL-6 and IL-8, MSCs increased the expression of the anti-inflammatory factor IDO during coculture with PBMCs. This shows the dual effect of MSCs on PBMC proliferation, which was further demonstrated by the persisted and even accelerated proliferation of PBMCs after removal of MSCs. This extra proliferative effect on PBMCs was not observed when PBMCs were kept in the presence of MSCs, suggesting a blockade of PBMC activation and proliferation. Taken together, these results indicate that MSCs have a regulatory function to maintain homeostasis in the (local) environment: under inflammatory conditions, MSCs are immunosuppressive, whereas MSCs stimulate T-cell proliferation under low inflammatory conditions.
The spectacular increase in the proliferation of PBMCs after removal of MSCs was for an overwhelming part dependent on the proliferation of CD4+ T-cells. These cells highly expressed CD25 and CD69, reflecting their activation status. Further, within the CD4+ T-cell population, the proportion of CD25posFoxP3+CD127neg Tregs was significantly increased. Our data therefore suggest that MSCs favor the expansion of Tregs. Functional analysis showed that the T-cells that expanded after removal of MSCs were not anergic and had immunosuppressive function. The CD25pos fraction of the generated cells was responsible for this effect, whereas the CD25neg fraction had no inhibitory effect on the proliferation of allo-activated cells. Expansion of these cells after removal of MSCs was crucial for obtaining this immunosuppressive capacity.
There is currently a trend to apply MSCs for an increasing number of applications, including organ transplantation. The relevance of the observed stimulatory effect of MSCs on resting T-cells in clinical therapy is unknown. Although the expansion of T-cells by MSCs harbors a potential danger, the net effect of MSCs was an enhancement of the regulatory capacity of the T-cell compartment. It is important for successful clinical application of MSCs that the interaction between MSC, Tregs, and other immune cells is studied in further detail under various immunological conditions.
In conclusion, the results of this study give new insights of the mechanisms of immune modulation by MSCs under noninflammatory conditions. Under these conditions, MSCs have a stimulating effect on the activation and proliferation of T-cells. At the same time, MSCs show immune regulatory functions that can control the full activation of T-cells. Removal of MSCs will lead to further activation and explosive proliferation of T-cells.
Footnotes
Acknowledgments
The authors thank the Department of Surgery of the Erasmus Medical Center Rotterdam for collecting the perirenal adipose tissue of the living kidney donors.
Author Disclosure Statement
No competing financial interests exist.