
Abstract
As granulocyte-colony-stimulating factor (G-CSF)-induced mobilization of hematopoietic stem/progenitor cells (HSPCs) increases human serum levels of hepatocyte growth factor (HGF), our aim was to investigate the role of HGF and its receptor, c-Met, in the mobilization of HSPC. CD34+ cells and leukocytes were isolated from the bone marrow (BM) of normal donors and the peripheral blood (PB) of patients mobilized with G-CSF and chemotherapy. Plasma HGF levels were evaluated by ELISA and HGF and c-Met expression by RT-PCR, fluorescence-activated cell sorter (FACS) analysis, and confocal microscopy. Because matrix metalloproteinases (MMPs) facilitate migration across extracellular matrix (ECM) and basement membranes, we also examined expression of MMP-9 and membrane type 1 (MT1)-MMP in hematopoietic cells after HGF stimulation. We found that plasma HGF levels in mobilized (m)PB were higher in patients who are good mobilizers and correlated with their white blood cell (WBC) and CD34+ cell counts. Moreover, HGF and c-Met expression was significantly higher in mPB CD34+ cells and leukocytes than in their steady-state BM counterpart cells and was up-regulated by G-CSF. Like G-CSF, HGF increased the secretion of MMP-9 and the expression of MT1-MMP in leukocytes, which was abrogated by the c-Met inhibitor K-252a. This inhibitor also significantly reduced the trans-Matrigel migration of mPB CD34+ cells toward HGF. Our results suggest that G-CSF-mediated HSPC mobilization occurs in part through the HGF/c-Met axis in HSPC and myeloid cells, eliciting increased production of matrix-degrading enzymes and subsequently facilitating egress of HSPC.
Introduction
H
We and others have reported that the HGF/c-Met axis is involved in the migration of mesenchymal stem cells, monocytes, and various tumor cells including rhabdomyo-sarcoma (RMS) cells by up-regulating matrix metallopro-teinase (MMP) secretion, especially of MMP-2 and MMP-9 [14 –18]. MMPs are known to play a key role in normal and malignant cell migration and invasion [19]. Following G-CSF administration, activated neutrophils release many pro-teolytic enzymes such as MMP-9 to the BM microenviron-ment, facilitating the egress of HSPC from the BM. G-CSF also up-regulates MMP-2 and MMP-9 in BM CD34+ cell progenitors, which under steady-state conditions do not secrete them [20]. Moreover, G-CSF up-regulates membrane type (MT)1-MMP in CD34+ cells and leukocytes and MT1-MMP has been shown to be necessary for the migration of these cells [21 –23].
Here we hypothesized that G-CSF administration increases production of HGF by leukocytes and CD34+ cells, which further up-regulates MMP-9 and MT1-MMP expression, resulting in a highly proteolytic BM microenvironment that facilitates HSPC egress.
Materials and Methods
Patients, cells, and cultures
BM was obtained from unrelated donors with their informed consent in accordance with the guidelines approved by the University of Alberta Ethics Committee. Peripheral blood (PB) samples were obtained with their informed consent from patients diagnosed with non-Hodg-kin's lymphoma who had been mobilized with chemotherapy (cyclophosphamide 4,000 mg/m2 IV and etoposide 200 mg/m2 qd × 3) followed by G-CSF (Filgrastim 5 μg/kg BID; Amgen, Thousand Oaks, CA) and from normal donors. During mobilization, CD34+ cell and white blood cell (WBC) counts were monitored, and leukapheresis was carried out using the Cobe Spectra Apheresis System (COBE, Lakewood, CO). Patients were regarded as good mobilizers when their PB CD34+ cell count reached ≥50 cells/μL and poor mobilizers when it was <50 cells/μL on the day of leu-kapheresis. Light-density mononuclear (MNC) cells from BM and from leukapheresis products were obtained by cen-trifugation (using a 60% Percoll density gradient, 1.077 g/mL; Amersham, Uppsala, Sweden). CD34+ cell separation was carried out using the Miltenyi MidiMACS system using LS separation columns (Miltenyi Biotec, Auburn, CA) according to the manufacturer's instructions. The purity of the isolated CD34+ cells was higher than 95% as determined by fluorescence-activated cell sorter (FACS) analysis using a FACscan (Becton Dickinson, San Jose, CA). Polymorphonuclear cells (PMN) were isolated by centrifugation using Lympholyte-poly (Cedarlane, Burlington, ON, Canada). BM leukocytes were prepared by lysing red blood cells with lysis buffer (150 mM NH4Cl, 1 mM EDTA, 10 mM NaHCO3) for 10 min at room temperature. For some experiments, leukocytes were stimulated with either 100 ng/mL G-CSF (R&D Systems, Minneapolis, MN) or 20 ng/mL HGF (PeproTech Inc, Rocky Hill, NJ) in RMPI 1640 with 5% fetal calf serum (FCS) for 48 h (37°C, 95% humidity, 5% CO2).
Human umbilical vein endothelial cells (HUVEC) were cultured on gelatin-coated flasks in media consisting of M199, 10 mM
ELISA
The level of HGF in plasma and in media conditioned by G-CSF-stimulated cells was measured by ELISA (RayBiotech, Inc., Norcross, GA) according to the manufacturer's instructions. Plasma was obtained from BM samples harvested from normal unrelated donors and from the PB plasma of mobilized patients on the day of leukapheresis, and stored at −20°C until use.
Expansion of progenitor cells
Myeloid and megakaryocytic progenitors were expanded from CD34+ cells as we described previously [25]. In brief, mPB CD34+ cells were cultured in XVIVO 20 media (BioWhittaker, Walkersville, MD) at 37°C in a fully humidified atmosphere supplemented with 5% CO2 for 6 days. Media were supplemented with recombinant human (rh) IL-3 (10 ng/mL) and rh GM-CSF (5 ng/mL) for CFU-GM expansion, and with rh thrombopoietin (TPO; 50 ng/mL) and rh IL-3 (10 ng/mL) for CFU-Meg progenitor expansion (all cytokines were obtained from PeproTech). The expanded cells were stained for CD34 and megakaryocytic lineage marker (CD41) and myeloid lineage marker (CD33) before use. The progenitor cells were stimulated with 100 ng/mL G-CSF for 48 h before FACS analysis was carried out.
Semiquantitative RT-PCR and densitometric analysis
Total RNA from CD34+ cells, leukocytes, HUVEC, and CFU-F was isolated using TRIZOL (Gibco-BRL, Gaithersburg, MD) as previously described [22]. To evaluate HGF and c-Met transcripts, RT-PCRs were carried out using primer sequences for human GAPDH (housekeeping gene), c-Met, HGF, and MT1-MMP as described previously [14,18]. Thermocycling was performed with an Eppendorf Mastercycler (Westbury, NY) and the PCR products were electrophoresed on a 2% agarose gel containing ethidium bromide. Gels were visualized under UV light and photographed using the Alpha Innotech Imaging System (San Leandro, CA). Densitometric analysis of the HGF, c-Met, MT1-MMP, and GAPDH bands in each sample was carried out using NIH Image J software (Bethesda, MD). The relative level of target mRNA was regarded as the ratio between the intensities of the HGF or c-Met and the GAPDH bands.
FACS analysis and confocal analysis
To evaluate c-Met expression cells were labeled with anti-c-Met MoAb, clone DO-24 (UPS Biotechnology, Lake Placid, NY), followed by staining with goat anti-mouse AlexaFluor-488 secondary antibody (Invitrogen). To evaluate MT1-MMP expression cells were labeled with anti-MT1-MMP (Chemicon, Billerica, MA) followed by staining with goat anti-rabbit AlexaFluor-555 (Invitrogen). In brief, the cells were washed 3 times in buffer (PBS/0.1% BSA) and incubated with isotype IgG, anti-c-Met MoAb or anti-MT1-MMP for 45 min on ice. Cells were washed 3 times and stained with secondary antibody for 30 min on ice. After the final wash, cells were fixed in 1% paraformaldehyde and analyzed by FACS. Lymphocytes, monocytes, and granulocytes were defined based on their forward scatter (FSC) and side scatter (SSC) using FCS Express (De Novo Software, Los Angeles, CA). Coverslips coated with fibronectin (10 pg/mL) were left overnight and washed. CD34+ cells were plated on the coverslips for 1 h and, after fixation with 3.7% paraformaldehyde, blocked with 1% BSA (for 30 min at room temperature). The cells were labeled with anti-c-Met MoAb overnight, washed 3 times, stained with goat anti-mouse AlexaFluor-488 for 45 min on ice, washed, and then DAPI nuclear stain (Invitrogen) was added before mounting in ProLong anti-fade reagent (Invitrogen). Slides were examined using an LSM510 laser scanning confocal microscope (Carl Zeiss, Gottingen, West Germany).
Zymography and trans-Matrigel migration
For zymography analysis, BM leukocytes (2 × 106/mL) were incubated in serum-free IMDM in the absence (control) or presence of HGF (20 ng/mL) or G-CSF (100 ng/mL) or G-CSF with 1 μM c-Met inhibitor, K-252a (Cedarlane), at 37°C in a humidified atmosphere supplemented with 5% CO2. After 48 h the cell-conditioned media were collected and, to determine MMP activity, MMP-2 and MMP-9 were analyzed by zymography, as we previously described [20]. Band intensity was evaluated by densitometry using the Alpha Innotech Imaging System.
The trans-Matrigel migration assay was carried out as we described in detail previously [20]. Polycarbonate filters were coated with the reconstituted basement membrane Matrigel (BD, Franklin Lakes, NJ). Prewarmed serum-free medium (IMDM/0.1% BSA) without (control) or with HGF (20 ng/mL) was placed in the lower chambers. The mPB CD34+ cells were preincubated with 1 μM K-252a (Cedarlane) at 37°C for 30 min and aliquots of 2 × 105 cells in 200 μL were loaded onto the upper chambers and incubated (37°C, 95% humidity, 5% CO2) for 3 h. The trans-Matrigel migration index was calculated as the ratio of the number of cells migrating across Matrigel toward an HGF gradient to the number of cells migrating toward media alone.
Statistical analysis
The correlations between HGF plasma levels (pg/mL) and CD34+ cell and WBC counts were analyzed using linear regression. Correlation coefficient values (R) > 0.5 were considered significant. Arithmetic means and standard deviations were calculated and statistical significance was defined as P ≤ 0.05 using Student's t-test.
Results
HGF levels are elevated in the plasma of good mobilizers
It has been previously reported that serum HGF levels are elevated during G-CSF-induced HSPC mobilization [3]. To elucidate the role of HGF in mobilization, we compared HGF levels in steady-state (ss)BM and PB plasma samples from unmobilized donors and mobilized patients. In steady state, HGF levels were significantly higher in BM than in PB plasma (Fig. 1A). However, in patients undergoing mobilization (with G-CSF and chemotherapy) the HGF levels were significantly higher in the plasma of patients who were identified as good mobilizers than in that of poor mobilizers (Fig. 1A). As neutrophilia precedes HSPC mobilization and the WBC count, in addition to the CD34+ cell count at the time of leukapheresis is considered to be predictive of the level of mobilization, accordingly we found significant correlation between the HGF plasma level and WBC count (R = 0.83, P = 0.0014, Fig. 1B) as well as CD34+ cell count (R = 0.8, P = 0.006, Fig. 1C) in the PB of mobilized patients. Consistent with these in vivo findings, we also found that the HGF level increased about 2-fold in media conditioned by BM mononuclear cells stimulated in vitro with G-CSF, as compared to unstimulated controls (Fig. 1D).
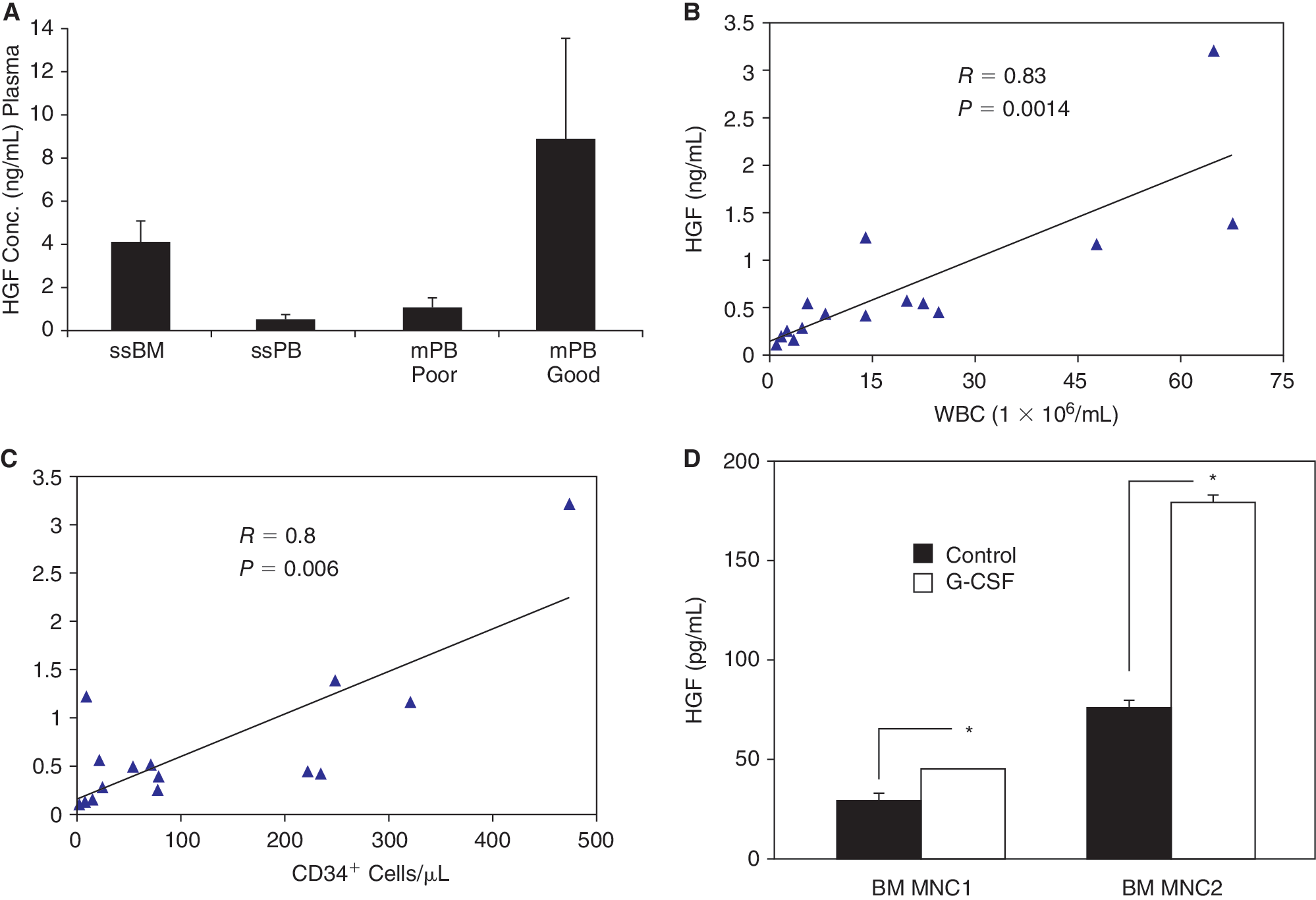
Hepatocyte growth factor (HGF) levels in plasma of donors, mobilized patients, and granulocyte-colony-stimulating factor (G-CSF)-treated bone marrow (BM) cells. (
G-CSF up-regulates HGF expression in CD34+ cells and leukocytes but not in stromal cells
We then examined whether there are differences in expression of HGF in cells derived from ssBM and ssPB versus mobilized (m)PB. As we had previously reported that the HGF transcript is expressed in ssBM and mPB CD34+ cells [25], we first confirmed these findings, but found in addition that HGF mRNA expression was significantly higher in mPB than in ssBM CD34+ cells. Moreover, it was also higher in mPB MNC and mPB PMN than in their nonmobilized counterpart cells (Fig. 2A). Consistent with this pattern of expression, when we preincubated nonmobilized, normal (n)BM MNC and leukocytes with G-CSF, we observed significant (P < 0.05) up-regulation of HGF expression (Fig. 2B). In contrast, G-CSF down-regulated HGF expression in BM stromal cells, and had no effect on HUVEC, which do not express HGF (Fig. 2B).
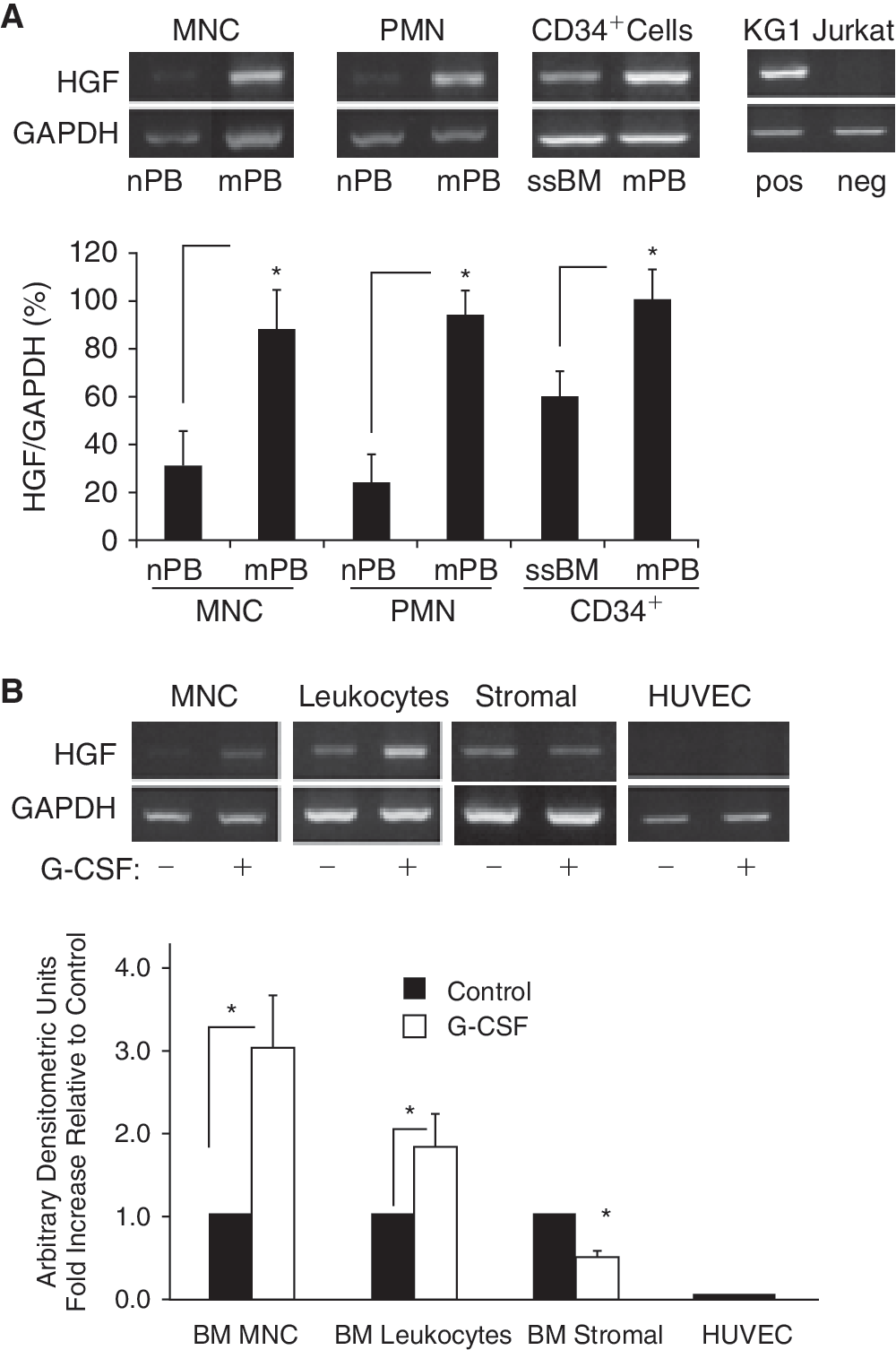
Hepatocyte growth factor (HGF) expression in CD34+cells and leukocytes. (
G-CSF up-regulates c-Met expression
Having shown a correlation between HGF levels and degree of mobilization in vivo and a stimulatory effect of G-CSF on HGF expression in vitro, we then examined various cells most likely responding to HGF signaling. Using flow cytometry, we evaluated the expression of the HGF receptor c-Met on normal, ssPB leukocytes (lymphocytes, monocytes, and PMN), and their mPB counterpart cells, and found that c-Met was not expressed on lymphocytes from either population. However, c-Met was expressed moderately on monocytes (38% ± 11%) and albeit weakly on PMN (10% ± 1.7%) from normal ssPB but more highly on monocytes (71.5% ± 8%) and PMN (79% ± 10%) from mPB (Fig. 3A). It had previously been demonstrated that ssBM CD34+ cells do not express c-Met [4] and here we observed using flow cytometry c-Met expression on <3% of ssBM CD34+ cells but on >30% of mPB CD34+ cells (Fig. 3B). Strong c-Met expression on mPB CD34+ cells was further confirmed by confocal microscopy (Fig. 3C).
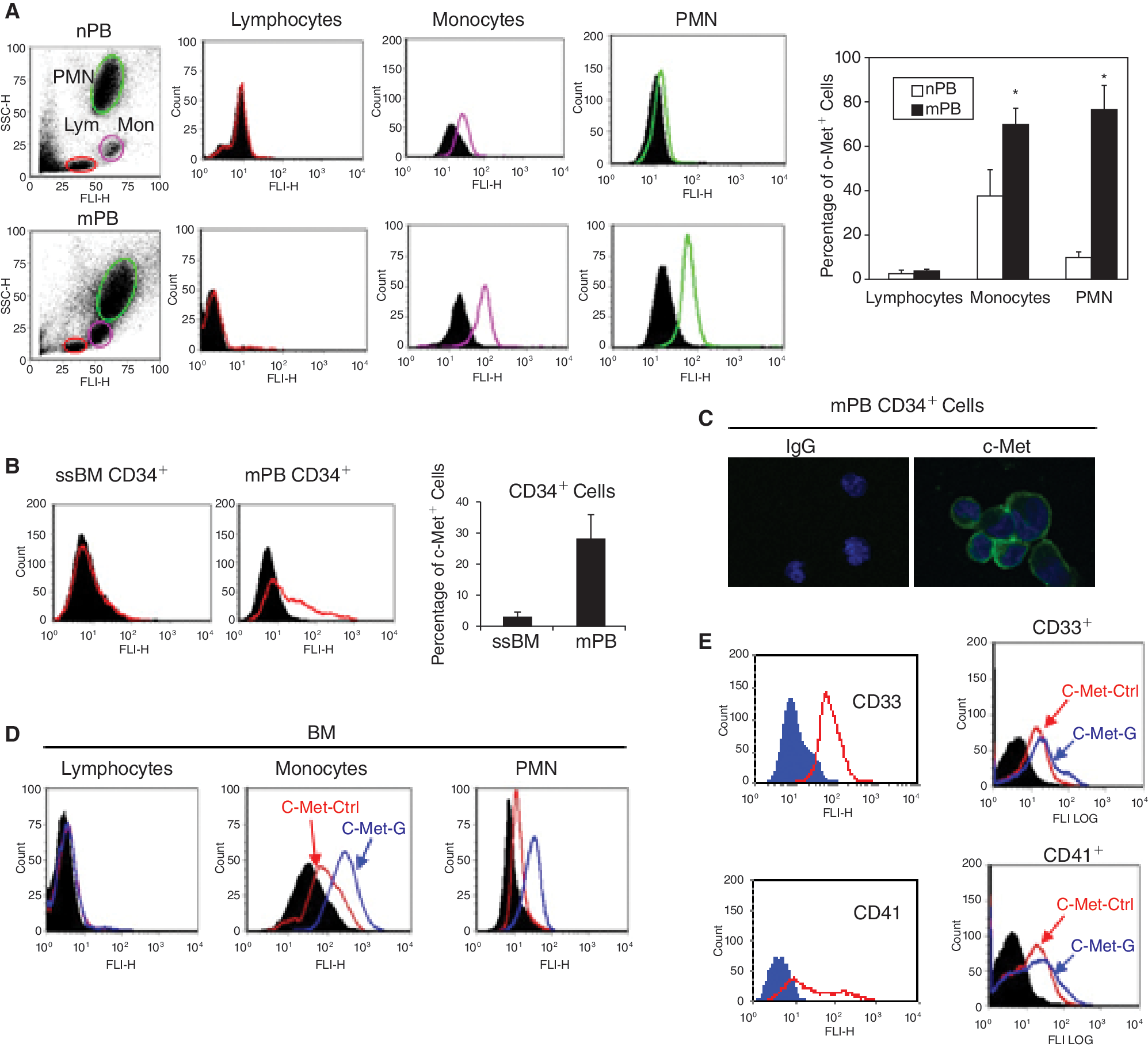
Fluorescence-activated cell sorter (FACS) analysis of c-Met in leukocyte subsets and CD34+ cells and the effect of granulocyte-colony-stimulating factor (G-CSF). (
Moreover, when normal BM leukocytes (monocytes and PMN) were preincubated with G-CSF, we observed an increase in c-Met expression (Fig. 3D). We also examined c-Met expression in CFU-GM and CFU-Meg progenitor cells (ex vivo expanded from mPB CD34+ cells) and found that G-CSF also up-regulates it in subpopulations (CD33+ and CD41+ cells, respectively) of these cells (Fig. 3E).
HGF, like G-CSF, increases MMP expression, which is inhibited by K-252a
G-CSF is known to stimulate the release of MMP-9 from neutrophils or HSPC [2,19], and we have reported that MMP-2 and MMP-9 secretion is up-regulated by HGF in RMS cells [18]. To examine whether HGF affects MMP expression in he-matopoietic cells, steady-state BM leukocytes were first stimulated with HGF. The cell-conditioned media were collected and evaluated for MMPs secretion by zymography, and cells were analyzed for MT1-MMP surface expression by flow cytometry. We found that HGF slightly increased MMP-9 secretion by these cells, and in the presence of the c-Met inhibitor, K-252a, MMP-9 secretion was reduced (Fig. 4A, left panels). When the leukocytes were gated by FACS (as in Fig. 3A), we found that HGF increased MT1-MMP surface expression on BM monocytes and PMN cells (Fig. 4B, top panel). Interestingly, we found that MMP-9 secretion, which is stimulated by G-CSF, can be abrogated by the c-Met blocking agent K-252a (Fig. 4A, right panels). Similarly, surface expression of MT1-MMP in BM monocytes and PMN cells, which was enhanced by treatment with G-CSF, was diminished in the presence of K-252a (Fig. 4B, bottom panels). Consistently, MT1-MMP gene expression was enhanced in BM MNC after treatment with G-CSF or HGF, which was almost obliterated in the presence of K-252a (Fig. 4C). This suggests that G-CSF-enhanced MMP-9 and MT1-MMP expression may occur partly through induction of the HGF/c-Met axis.
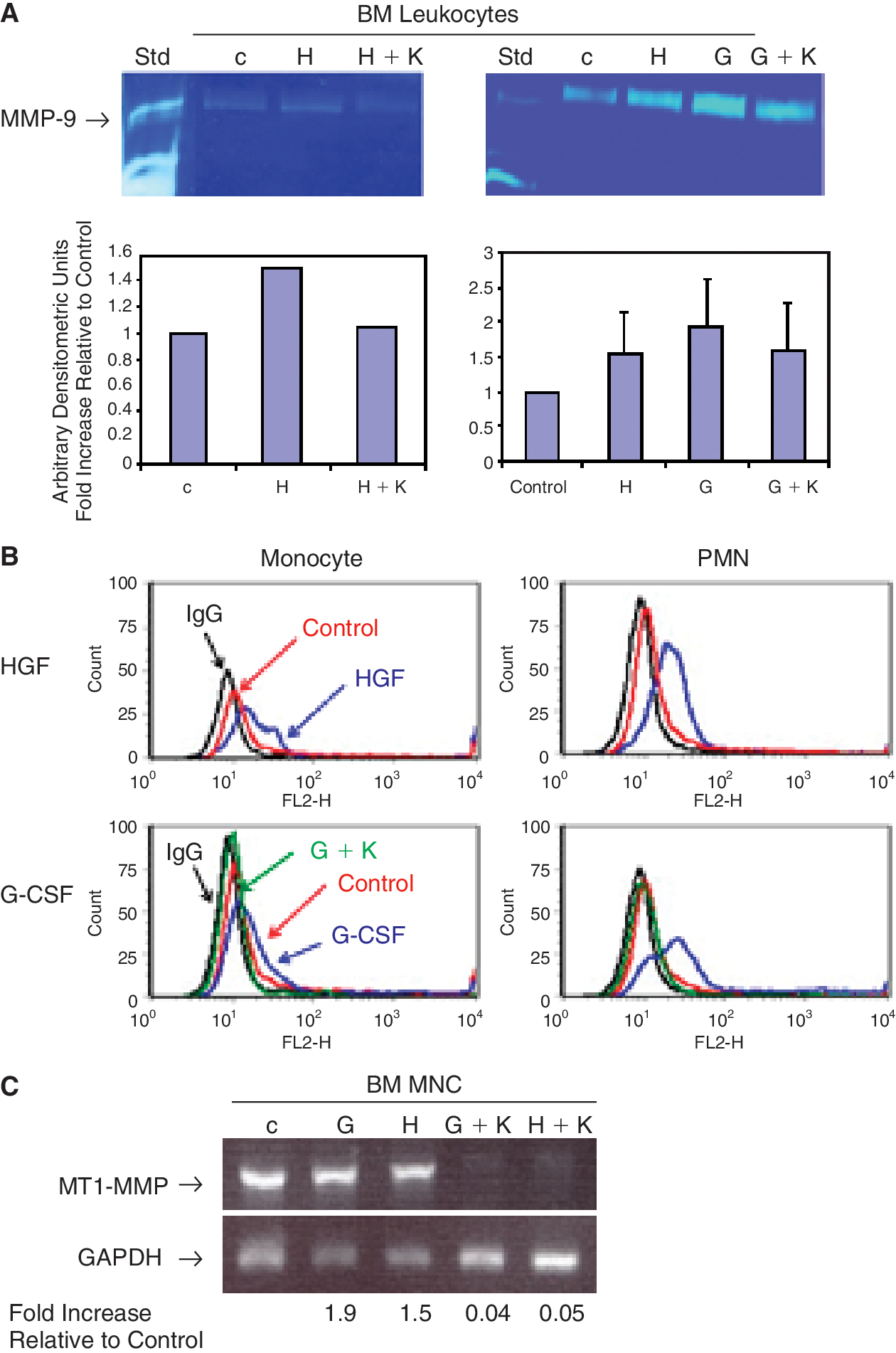
Hepatocyte growth factor (HGF) and granulocyte-colony-stimulating factor (G-CSF) up-regulates expression of MMP-9 and MT1-MMP in BM leukocytes, which was abrogated by c-Met antagonist. (
K-252a inhibits trans-Matrigel invasion of mPB CD34+ cells toward HGF
As mPB CD34+ cells showed significantly enhanced expression of c-Met (Fig. 3B and 3C), we evaluated whether an HGF gradient increases their chemoinvasion. We found they showed increased transmigration across Matrigel toward an HGF gradient compared to the control, and this was abrogated by K-252a (Fig. 5A).
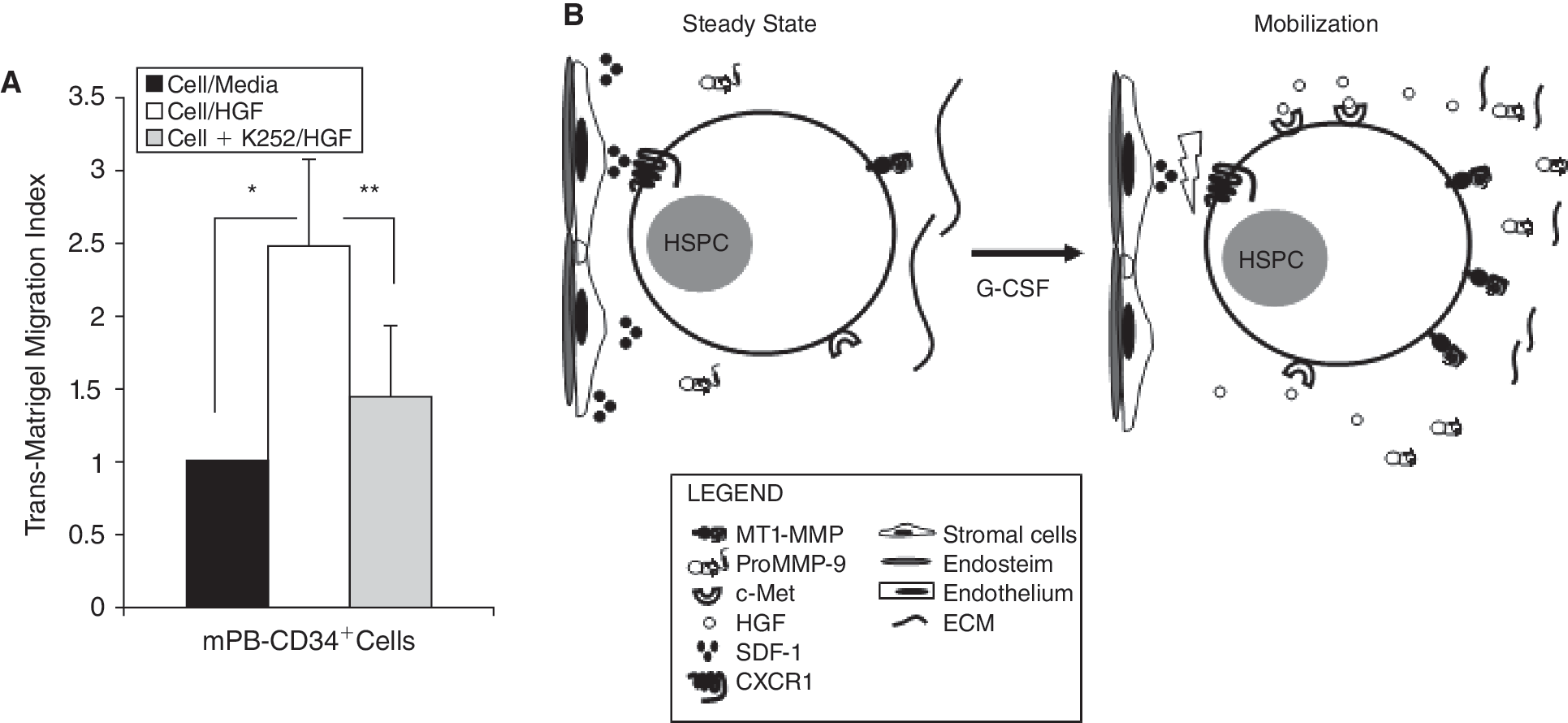
c-Met antagonist inhibits trans-Matrigel migration of granulocyte-colony-stimulating factor (G-CSF)-mobilized hematopoietic stem/progenitor cell (HSPC) toward hepatocyte growth factor (HGF): crosstalk between G-CSF and HGF/c-Met axis in HSPC mobilization. (
Discussion
Although the HGF/c-Met axis is known to play an important role in cell trafficking [5,17], its role in HSPC mobilization has not been fully established. HGF is produced in the BM microenvironment and also exists in neutrophil granules that release it upon neutrophil activation [13]. HGF serum levels were found to be elevated in donors mobilized with G-CSF [3] and in this work we found elevated levels of HGF in the plasma of patients treated with G-CSF. These levels correlated with the number of CD34+ cells and WBC in patients' PB, and were very low in patients who were poor mobilizers. As transient neutrophilia and leukocytosis are commonly observed during G-CSF-induced mobilization [2], the most likely sources of HGF in the plasma of G-CSF-mobilized patients are activated leukocytes; however, other sources cannot be excluded. Moreover, we demonstrated that in mobilized leukocytes and mPB CD34+ cells HGF expression is significantly higher than in their steady-state counterpart cells. Furthermore, in vitro G-CSF stimulation of ssBM leukocytes up-regulated HGF at the mRNA and protein levels, again consistent with the elevated HGF levels found in mobilized plasma. It is conceivable that in patients, chemotherapy, as well as in disease stage, has an impact on the HGF/c-Met axis so it would be interesting in the future to also evaluate samples from donors mobilized with G-CSF for allogeneic transplantation.
We also report here for the first time that c-Met is highly expressed on mPB CD34+ cells but not on ssBM CD34+ cells, indicating that its expression occurs on more mobile HSPC. This finding accords with the known involvement of the HGF/c-Met axis in the migration/metastasis of tumors such as RMS [18], breast, and liver [17], and of leukemic blasts [26], as well as in the migration of mesenchymal stem cells [14]. Moreover, activated monocytes have been shown to express c-Met [12], which agrees with our findings here that G-CSF-stimulated leukocytes highly express c-Met, and with a report that they occur in relative abundance in G-CSF-mobilized PB [27].
SDF-1, produced by the BM stromal cells and osteoblasts, plays a crucial role in the retention of HSPC within the BM via its interaction with the CXCR4 receptor expressed by HSPC. However, during G-CSF mobilization SDF-1 is inactivated by the proteolytic enzymes secreted by activated neutrophils, leading to egress of HSPC [1,2]. HGF is also constitutively produced by BM stromal cells [6] and has been shown to stimulate their production of SDF-1a [9]. On the other hand, addition of neutralizing anti-HGF antibody to BM stromal cells was shown to reduce their production of SDF-1α, as determined by ELISA, in a dose-dependent manner, which was subsequently restored by the addition of recombinant HGF. Moreover, transcription of SDF-1a was also inhibited by HGF neutralizing antibody [9]. Here we found that G-CSF treatment significantly down-regulates HGF expression by BM stromal cells, indicating that G-CSF, through this mechanism, may indirectly reduce production of SDF-1 in the BM microenviron-ment, which could facilitate the mobilization of HSPC [2].
Furthermore, proteolytic enzymes such as MMPs are widely involved in the migration of normal and malignant cells by degrading most extracellular matrix (ECM) components as well as numerous non-ECM proteins [19,28]. Previously, we reported that both MMP-2 and MMP-9 are secreted by mPB CD34+ cells, but not by ssBM CD34+ cells, and we postulated a role for MMPs in HSPC mobilization [20]. Here, we demonstrate that both HGF and G-CSF increase MMP-9 secretion by BM leukocytes and this secretion is reduced by a c-Met inhibitor. Another study demonstrated that all these factors (HGF, SDF-1, and MMP-9) are also involved in stress-induced migration of BM CD34+ cells to the liver [29]. Moreover, we and others recently observed that a membrane-bound MMP, MT1-MMP, also plays a crucial role in G-CSF-induced HSPC mobilization [21,23] and in the migration of monocytes [30]. MT1-MMP displays a wide range of proteolytic activities. It not only degrades ECM components such as gelatin, fibronectin, laminin, vitronectin, and fibrillar collagens, but also activates proMMP-2 and proMMP-9, inactivates SDF-1, cleaves CD44 and tissue transglutaminase, and processes various growth factors and cytokines [28]. Here we demonstrated that both HGF and G-CSF up-regulated the expression of MT1-MMP, which could result in a highly proteolytic microenvironment in the BM and contribute to the subsequent release of HSPC to the PB. Based on our results, we can speculate on a possible crosstalk between G-CSF and the HGF/c-Met axis; however, elucidation of the exact mechanism will require further study.
In summary, we envision that during G-CSF-induced mobilization (i) there is an increase in the HGF plasma levels in circulating blood (which we showed to be significantly higher in patients who are good mobilizers), with a concomitant decrease in the production of HGF by BM stromal cells and (ii) production of SDF-1 is reduced within the BM at the same time as it is inactivated by elevated MMP-9 and MT1-MMP. Moreover, during mobilization G-CSF also enhances the expression of c-Met in myeloid cells and HSPC in the BM, rendering them more responsive to elevated HGF levels in the PB plasma, promoting their egress. Furthermore, G-CSF, together with HGF, increases secretion of MMPs in the BM and MT1-MMP at the cell surface, facilitating pericellular ECM prote-olysis that contributes to HSPC mobilization (Fig. 5B). Thus, the HGF/c-Met axis could possibly synergize with G-CSF in providing directional cues for the mobilization of HSPC from the BM to the circulation. The development of agents to target this axis could improve mobilization and allow for more efficient collection of HSPC for transplantation.
Footnotes
Acknowledgments
This work was supported by grants from Canadian Blood Services (CBS) R&D/Canadian Institutes of Health Research (CIHR), Blood Utilization and Conservation Initiative to AJ-W and a CBS Postdoctoral Fellowship award to AJ. We thank Sara Ilnitsky, Jencet Montano, April Xu, and Barbara Pedrycz for technical help.
Author Disclosure Statement
No competing financial interests exist.