
Abstract
Due to their immunosuppressive properties, human mesenchymal stem cells (hMSC) represent a promising tool for cell-based therapies of autoimmune diseases such as multiple sclerosis (MS). Mouse MSC (mMSC) have been used extensively to characterize and optimize route of administration, motility, cellular targets, and immunosuppressive mechanisms in mouse models of autoimmune diseases, such as experimental autoimmune encephalomyelitis (EAE). Tryptophan (trp) catabolism by indolamine-2,3-dioxygenase 1 (IDO1) is a chief endogenous metabolic pathway that tightly regulates unwanted immune responses through depletion of trp and generation of immunosuppressive kynurenines (kyn). IDO1 activity contributes to the immunosuppressive phenotype of hMSC. Here, we demonstrate that although IDO1 is inducible in bone marrow-derived mMSC by proinflammatory stimuli such as interferon-g (IFN-g) and ligands of toll-like receptors (TLR), it does not lead to catabolism of trp in vitro. This failure to catabolize trp is not due to defective TLR signaling as demonstrated by induction of interleukin 6 (IL-6) by TLR activation. While mMSC suppressed the activation of antigen-specific myelin oligodendrocyte glycoprotein (MOG)-reactive T-cell receptor (TCR) transgenic T-helper (TH) cells in co-culture, neither pharmacologic inhibition nor genetic ablation of IDO1 reversed this suppressive effect. Finally, systemic administration of both, IDO1-proficient and phenotypically identical IDO1-deficient mMSC, equally resulted in amelioration of EAE. mMSC, unlike hMSC, do not display IDO1-mediated suppression of antigen-specific T-cell responses.
Introduction
M
Materials and Methods
Mice
C57Bl/6 and SJL mice were purchased from Charles River Laboratories (Sulzfeld, Germany). Transgenic IDO1-deficient (IDO1−/−) mice were generated as previously described [26] and were backcrossed on a C57Bl/6 background. 2D2 mice on a C57Bl/6 background expressing a T-cell receptor (TCR) specific for the myelin oligodendrocyte glycoprotein (MOG) peptide p35–55 were kindly provided by Vijay Kuchroo [27]. The transgenic TCR in heterozygous mice was detected by flow cytometry of murine blood using antibodies against CD4 and Vβ11 (BD Pharmingen, Heidelberg, Germany). All animal work was performed in accordance with the German animal protection law under the permission of the local authorities in Tübingen and the National Institutes of Health guidelines Guide for the Care and Use of Laboratory Animals.
MSC preparation
MSC were isolated from C57Bl/6, IDO1−/−, and SJL mice. Femurs and tibiae were removed and cleaned of muscles and connective tissue. Epiphyses were cut off and bone marrow cells were flushed out with D-MEM (HyClone, Logan, UT) using a 23-gauge needle. Cells were centrifuged at 1,200 rpm and 4°C for 10 min, suspended in Murine MesenCult® Basal Medium for Mouse Mesenchymal Stem Cells (Stem Cell Technologies, Cologne, Germany) containing 20% Mouse Mesenchymal Stem Cell Stimulatory Supplement (Stem Cell Technologies), 100 U/mL penicillin, and 100 μg/mL streptomycin (Cambrex, Charles City, IA) (complete Murine MesenCult® Medium), plated in 75-cm2 flasks (Corning, Corning, NY) at a concentration of 5 × 105/mL and incubated at 37°C and 5% CO2. Medium was changed after 7 days and the adherent cell fraction expanded in complete Murine MesenCult® Medium. MSC were characterized by flow cytometry using a DAKO Cytomation CyanADP Analyzer (DAKO Cytomation, Hamburg, Germany) and DAKO Summit software. The following antibodies were used for FACS analysis: anti-CD11b, anti-CD34, anti-CD44, anti-CD45 (ebioscience, San Diego, CA), anti-CD9 (BD Pharmingen), anti-CD29 (Biolegend, San Diego, CA), anti-CD90 (Cedarlane, Burlington, ONT, Canada), and corresponding isotype controls (ebioscience). Osteogenic differentiation was performed as described [28] with minor variations. In brief, osteogenic differentiation was induced by adding 0.1 mM dexamethasone, 0.05 mM ascorbic acid, and 10 mM glycerol 2-phosphate disodium salt hydrate (Sigma, Munich, Germany) to complete D-MEM, containing 10 % fetal calf serum (FCS; Biochrom, Berlin, Germany), and 100 U/mL penicillin and 100 μg/mL streptomycin (PAA Laboratories, Pasching, Austria). Cells were plated in this medium in 94-mm tissue culture dishes (Cellstar/Brightpoint, Trier, Germany). Control cells were seeded in complete D-MEM without further supplements. After 6 weeks of culture, calcium deposits were detected by Alizarin Red S (Sigma) staining.
Dendritic cell (DC) preparation
Dendritic cell (DC) were generated from C57Bl/6 and SJL mice. Bone marrow cells were isolated as described above. Cells were suspended in RPMI 1640 (Cambrex), supplemented with 10% FCS (Biochrom), 100 U/mL penicillin, and 100 μg/mL streptomycin (Cambrex; complete RPMI), and 20 ng/mL (106 U/mL) recombinant murine Granulocyte Macrophage Colony-Stimulating Factor (rm-GM-CSF) (Immunotools, Friesoythe, Germany). The 107 bone marrow cells were seeded in 75-cm2 flasks (Corning). On day 2, 10 mL of complete RPMI, containing 40 ng/mL rm-GM-CSF, was added. On day 4, cells were washed twice with PBS and detached by incubating cells with 3 mL of accutase for 15 min at 37°C. Cells were centrifuged at 1,200 rpm and 4°C for 10 min, then resuspended in complete RPMI containing 20 ng/mL rm-GM-CSF at 5 × 105 cells/mL and seeded in 6-well plates with 3 mL/well. On day 6, 3 mL of complete RPMI, supplemented with 40 ng/mL rm-GM-CSF, was added. On day 7, we obtained immature DC, ready to be matured via cytokines or TLR ligands.
T-cell proliferation assay
For T-cell proliferation assays, spleens were removed and dissociated through a 40-μm cell strainer (BD Falcon, Heidelberg, Germany) to singularize splenocytes and to remove connective tissue, then washed with PBS and centrifuged at 1,200 rpm and 4°C for 10 min. Lysis of erythrocytes was performed by suspending the pelleted splenocytes in ACK Lysing Buffer (Cambrex) for 1 min. Cells were washed twice with PBS and resuspended in complete RPMI. For T-cell proliferation assays, splenocytes were seeded in 96-well plates (5 × 105 cells/well) on 0–5,000 mMSCs from C57Bl/6 or IDO−/− mice seeded 2 days before. Before 24 h, mMSCs had been pretreated with 50 μg/mL polyinosinic:polycytidylic acid (pI:C; Sigma), 5 μg/mL lipopolysaccharide (LPS, from Salmonella typhii; Sigma), or 500 IU/mL IFN-γ (Immunotools). Then mMSC were washed 3 times with PBS to remove all remnants of pI:C, LPS, and IFN-γ. Immediately, splenocytes (5 × 105 cells/well) of 2D2 mice were added. Activation of T cells was achieved by adding 10 μg/mL MOG p35–55 (MEVGWYRSPFSRVVHLYRNGK, Stanford Protein and Nucleic Acid Biotechnology Facility, Stanford, CA). In some experiments, 1 mM 1-methyl-
qRT-PCR
RNA was isolated using the Qiagen RNAeasy RNA isolation kit (Qiagen, Hilden, Germany) according to manufacturer's instructions. cDNA was synthesized with the Superscript™ Choice System (Invitrogen, Carlsbad, CA) using random hexamers. qRT-PCR was performed in 3 serial dilutions, each in duplicates using an ABI 7000 thermal cycler with SYBR Green PCR Mastermix (Eurogentec, Cologne, Germany). Data were evaluated with AB 7000 System SDS software. PCRs were checked by including controls without prior reverse transcription and by both melting curve and gel analysis. Relative quantification of gene expression was determined by comparison of threshold values. All results were normalized to b-actin. Primers were designed across exon boundaries. The sequences of the primers were the following: TLR1: fwd: TCAAGCATTTGGACCTCTCCT, rev: TTGTACCCGAGAACCGCTCA; TLR2: fwd: CCAGA CACTGGGGGTAACATC, rev: CGGATCGACTTTAGAC TTTGGG; TLR3: fwd: GGGGTCCAACTGGAGAACCT, rev: CCGGCGAGAACTCTTTAAGTGG; TLR4: fwd: ATGGC ATGGCTTACACCACC, rev: GAGGCCAATTTTGTCTCC ACA; TLR5: fwd: TCAGACGGCAGGATAGCCTTT, rev: AATGGTCAAGTTAGCATACTGGG; TLR6: fwd: GACTCT CCCACAACAGGATACG, rev: TCAGGTTGCCAAATTCCTT ACAC; TLR7: fwd: TCTTACCCTTACCATCAACCACA, rev: CCCCAGTAGAACAGGTACACA; TLR8: fwd: CAAACAA CAGCACCCAAATGAA, rev: AGGCAACCCAGCAGG TATAGT; TLR9: fwd: ACTCCGACTTCGTCCACCT, rev: GGCTCAATGGTCATGTGGCA; b-actin: fwd: TGTCCC TGTATGCCTCTGGT, rev: CACGCACGATTTCCCTCTC; IDO1: fwd: GCTTTGCTCTACCACATCCAC, rev: CAGG CGCTGTAACCTGTGT; KYNA: fwd: AGTGGGCTGCAC TTTTATACTG, rev: TGCAAACAGGTTGCCTTTCAG; KMO: fwd: TGATGTGTACGAAGCTAGGGA, rev: TCATGGGCA CACCTTTGGAAA; 3-HAAO: fwd: GGAGGCCCCAATACC AGGA, rev: TATAGGCACGTCCCGGTGTT.
Cytokine ELISA
Supernatants of mMSC for cytokine ELISA were generated by plating 5 × 104 cells in 24-well plates (BD Falcon) in 1 mL of complete RPMI. After 24 h, cells were stimulated with 50 μg/mL pI:C, 5 μg/mL LPS, or 500 IU/mL IFN-γ. Supernatants were collected after 24 and 72 h. ELISA for interleukin 2 (IL-2), IL-4, IL-6, IL-10, IL-12p40, IL-17A, IFN-γ, and tumor necrosis factor (TNF; ebioscience) were performed according to manufacturer's instructions.
Annexin–PI flow cytometry analysis
For Annexin- and propidium iodide (PI) staining, cells were seeded in 6-well plates at a concentration of 105/well in complete RPMI. After 24 h, cells were treated with 50 μg/mL pI:C or 5 μg/mL LPS for 24 h. Cells were detached using accutase, washed twice with PBS, and stained with Annexin V-FITC (BD Pharmingen) and PI (Sigma) for 15 min. Flow cytometry was performed using a Cyan ADP Analyzer (DAKO Cytomation) and DAKO Summit software.
HPLC analysis of tryptophan metabolites
Supernatants of mMSC were obtained as described above. HPLC analysis was performed according to Hervé et al. [29] using a Beckman HPLC with PDA detection and Lichrosorb RP-18 column (250 mm × 4 mm ID, 5 μm). Kyn release and tryptophan degradation were measured in complete RPMI 1640 media (Cambrex). The medium was harvested from 24-well plates at the indicated time points, centrifuged, and frozen until further analysis. After thawing, the samples were supplemented with trichloroacetic acid for protein precipitation, centrifuged, and 100 μL of the supernatant was analyzed by HPLC. Standard curves were generated with
Induction and treatment of EAE
Female C57Bl/6 mice were immunized subcutaneously (s.c.) with 100 μg/mouse MOG p35–55 in incomplete Freund's Adjuvants (Difco, Detroit, MI) containing 200 μg Mycobacterium tuberculosis (Difco) per mouse. Concomitantly, 200 ng/mouse pertussis toxin diluted in 100 μL of PBS was injected i.v., which was repeated 2 days later. Clinical signs of disease were scored daily according to a standard scoring system, representing 0 = no clinical signs, 1 = loss of tail tone, 2 = hind limb weakness, 3 = complete hind limb paralysis, 4 = hind limb and forelimb paralysis, 5 = moribund or dead. On days 4 and 9 after immunization, mice were injected intraperitoneally (i.p.) with 106 mMSC from C57Bl/6 or IDO1−/− mice, diluted in 200 μL of PBS. Control mice received equal volumes of PBS without cells.
Statistical analyses
Results were assessed by applying Student's t-test statistics to the experimental data obtained in vitro. In vivo data were statistically evaluated using the Mann–Whitney U-test.
Results
Characterization of mMSC
MSC were isolated from wild-type C57Bl/6, IDO−/− C57Bl/6 and SJL/J mice. Flow cytometry demonstrated that the cells were CD9+, CD29+, CD44+, CD90+, CD11b−, CD34−, CD45−, which is indicative of an MSC phenotype (Fig. 1A). Staining of calcium deposits verified the capacity to differentiate along the osteogenic lineage (Fig. 1B).
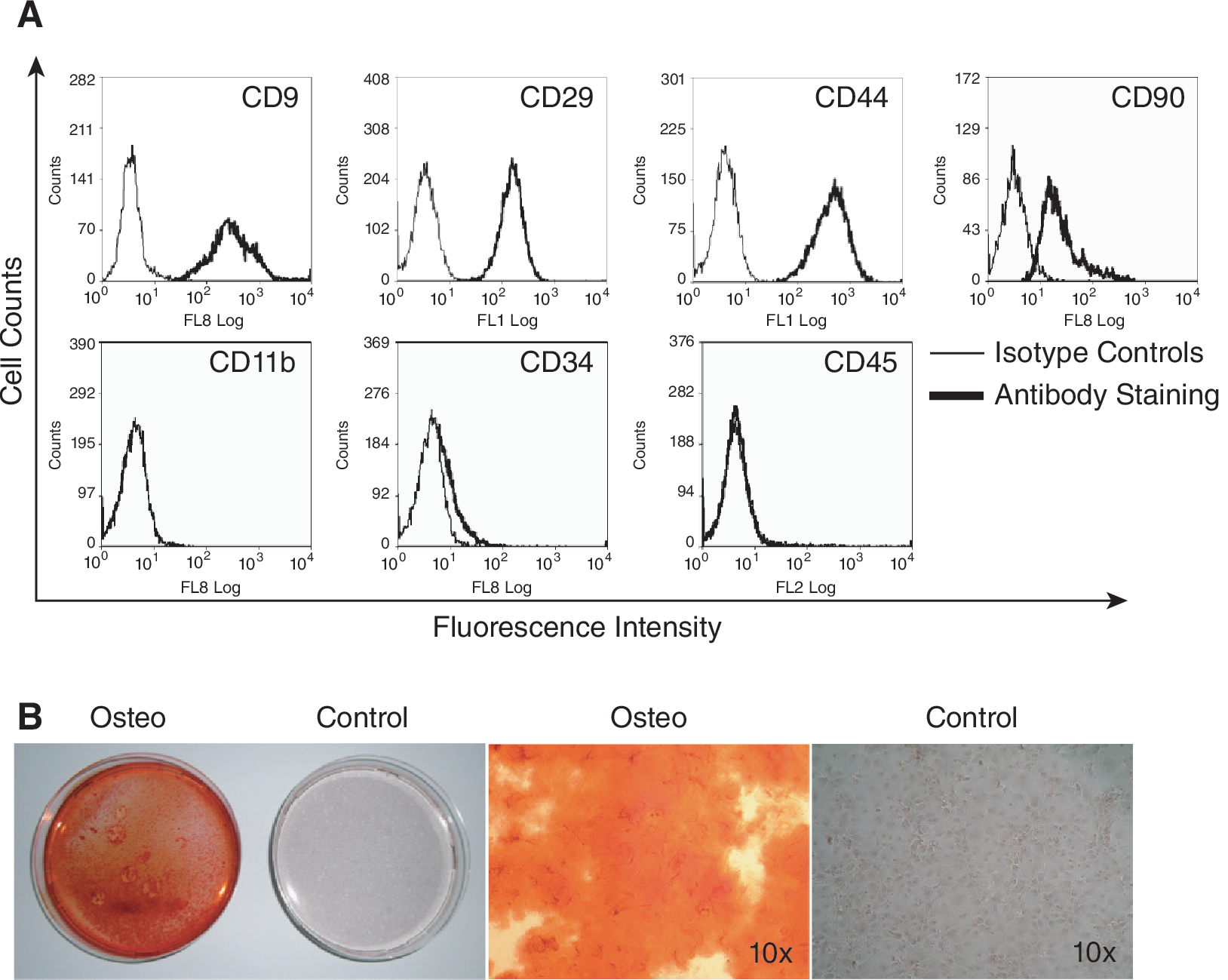
Characterization of mouse mesenchymal stem cells (mMSC). (
Tryptophan catabolism in mMSC
Trp catabolism was analyzed in mMSC. To this end, mMSC were stimulated with IFN-g or the TLR ligands pI:C or LPS. Then, mRNA expression of IDO1, the main inducible trp catabolizing enzyme, and the enzymes in its downstream pathway, kynureninase (KYNA), kynurenine monooxygenase (KMO), and 3-hydroxyanthranilic acid oxidase (3-HAAO) (Fig. 2) was determined. Naïve mouse DC served as a positive control. As opposed to mDC, there was no constitutive IDO1 mRNA expression in mMSC. While IFN-g induced a strong up-regulation in both mMSC and mDC, induction of IDO1 mRNA by TLR ligands was weak in mMSC (Fig. 3A).

Catabolic pathway of tryptophan (modified from Ref. [25]).
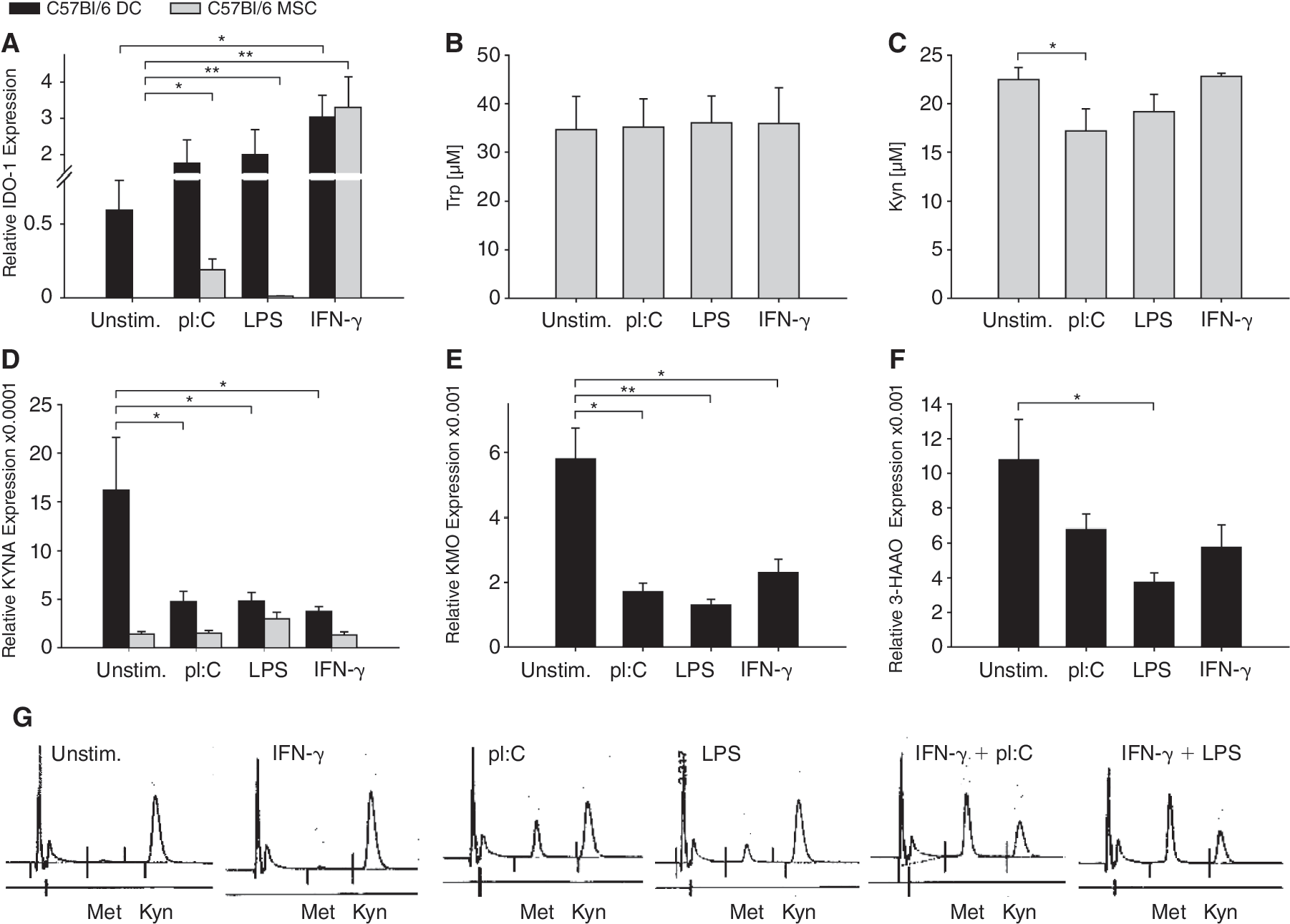
Effects of TLR ligands and IFN-γ on tryptophan-catabolizing enzymes. (
HPLC analysis of trp demonstrated that the increase of IDO1 mRNA in mMSC by TLR ligation or IFN-g stimulation did not result in IDO1 enzyme activity despite induction of IDO1 mRNA (Fig. 3B). Interestingly, kyn in the cell culture media decreased after TLR ligation (Fig. 3C), possibly indicating immediate catabolization via the downstream enzymes. We thus analyzed the expression of downstream kyn-catabolizing enzymes. KYNA was constitutively expressed in mDC and—albeit at lower levels—mMSC, but unlike in mDC it was not down-regulated in mMSC by TLR ligands or IFN-g (Fig. 3D). IFN-γ or TLR stimulation resulted in down-regulation of mRNA expression of KMO and 3-HAAO only in mDC, while mRNA of those 2 enzymes was not detected in mMSC (Fig. 3E and 3F). Interestingly, when mMSC were stimulated with pI:C or LPS, HPLC analysis detected an additional peak that does not appear after stimulation with IFN-g but can be increased by combination of TLR ligand and IFN-g. This additional peak correlated reciprocally with the peak size of kyn, suggesting it may be a derivative of it (Fig. 3G). This peak is not identical with trp and all its tested metabolites: kynurenic acid, 3-hydroxykynurenine, anthranilic acid, 3-hydroxyanthranilic acid, picolinic acid, quinolinic acid, kynuramic acid, or nicotinic acid.
mMSC express functional TLR
The reduced responsiveness of mMSC to TLR activation is in sharp contrast to hMSC, which display strong IDO1 activity in response to TLR ligation [19]. To see whether this reduced responsiveness reflects impaired functionality of TLR in mMSC, we first analyzed TLR mRNA expression. Figure 4A shows that both mDC and mMSC, isolated from C57Bl/6 mice, express TLR1–9. Expression levels of mDC and mMSC were comparable except for TLR7, TLR8, and TLR9, which were expressed at orders of magnitude lower in mMSC compared to mDC. TLR3 and TLR4, the receptors for pI:C and LPS, respectively, were highly expressed in mMSC. mMSC from SJL/J mice showed similar flow cytometric profile and similar TLR expression patterns as C57Bl/6 mMSC (Fig. 4B and 4C). To analyze the functionality of TLR3 and TLR4 in mMSC, we determined the mRNA expression and release of various cytokines in response to activation with pI:C and LPS. Both pI:C and to a lesser extent LPS induced the release of IL-6 in mMSC and mDC (Fig. 4D and 4E), indicating that the lack of relevant induction of trp catabolism by TLR ligands in mMSC is not due to lack of functional TLR signaling. There was no change in the production of the other tested cytokines: IL-10, IL-12, IL-23, IFN-γ, and TNF (Table 1). IL-2, IL-4, and IL-17 were not detected (data not shown). Of note, TLR activation did not result in mMSC death (Fig. 4F).

TLR expression on mouse mesenchymal stem cells (mMSC). (
Production of IL-6, IL-10, IL-12/23p40, IFN-γ, and TNF measured by ELISA in cultured DC, and IDO−/− MSC or wild-type MSC. Values are given as mean pg/mL from duplicates. Data are representative of 2 independent experiments.
Abbreviations: n/a: not applicable; MSC: mesenchymal stem cell.
mMSC suppress myelin-specific T cells
As an in vitro model for antigen-specific T-cell immunity, we chose myelin-specific T cells from 2D2 mice with a transgenic TCR specific for the immunodominant peptide p35–55 of MOG expressed on CD4+ T cells. MOG p35–55-specific IFN-g-secreting TH1-polarized T cells can be induced to become encephalitogenic in EAE [27]. IFN-g, in turn, induces IDO1 expression in mMSC (Fig. 3A). To evaluate whether mMSC suppress the proliferation of MOG-specific CD4+ T cells, splenocytes from 2D2 mice were activated with MOG p35–55 in the presence of syngeneic mMSC. Figure 5A shows that mMSC suppress the proliferation of MOG-specific CD4+ T cells in a concentration-dependent manner. Adding 1-l-MT, a competitive IDO1 inhibitor [19], to the mMSC/splenocyte culture did not restrict the capacity of mMSC to suppress T-cell proliferation indicating that mMSC-derived IDO1 is not involved in the suppression of T-cell proliferation (Fig. 5B). Of note, in the absence of mMSC, 1-
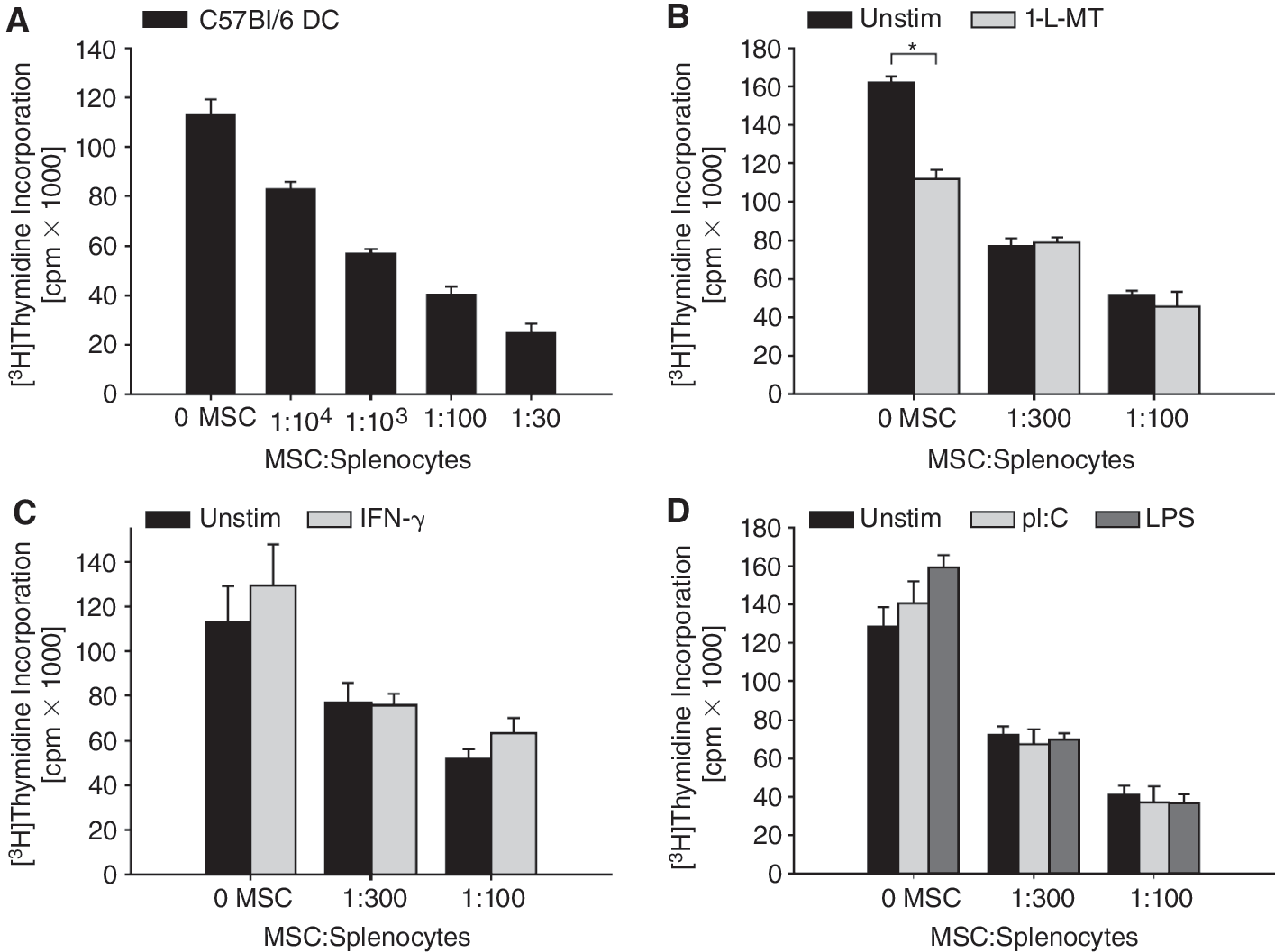
Indolamine-2,3-dioxygenase 1 (IDO1)-independent immunosuppression by mouse mesenchymal stem cell (mMSCs). (
IDO1 is not involved in mMSC-mediated immunosuppression in vitro
To further substantiate the finding that IDO1 is not involved in mMSC-mediated inhibition of MOG-specific T cells, we used mMSC from IDO1-deficient mice. IDO1−/−mMSC did not differ substantially from wild-type C57Bl/6 mMSC with respect to phenotype, differentiation capacity, and TLR expression (Fig. 6A–6D). IL-6 induction in response to LPS, however, was lower in IDO−/− MSC than wild-type MSC (Fig. 6D). When incubated with 2D2 splenocytes activated with MOG p35–55, IDO1−/− mMSC displayed similar immunosuppressive activity as their wild-type counterparts, indicating that IDO1 is not involved in mMSC-mediated modulation of TH cell activation (Fig. 6E).
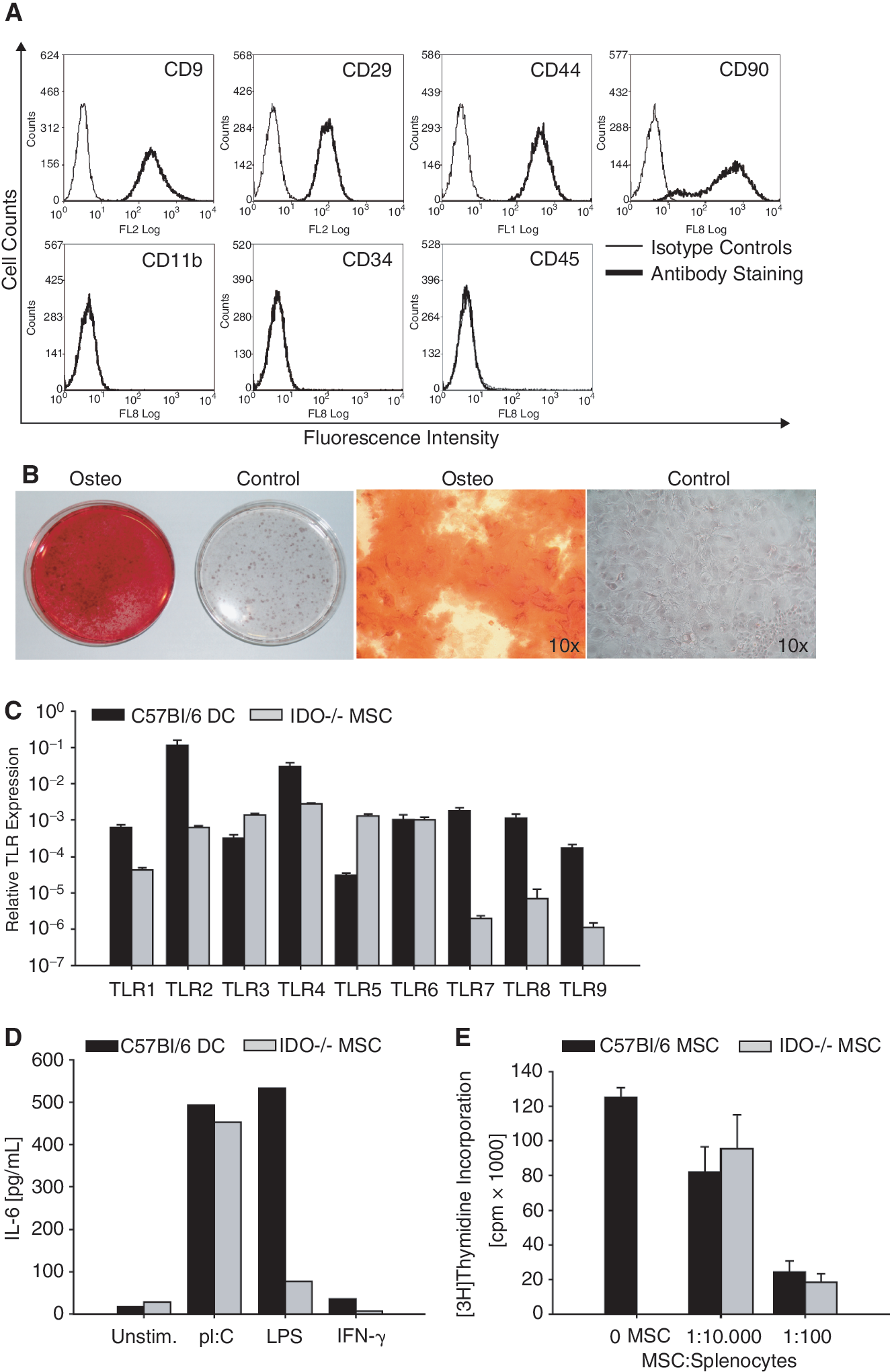
Indolamine-2, 3-dioxygenase 1 (IDO1) is dispensable for mouse mesenchymal stem cells (mMSCs)-mediated immunosuppression in vitro. (
IDO1 is not involved in mMSC-mediated immunosuppression in EAE
We next analyzed the role of mMSC trp catabolism in vivo by immunizing C57Bl/6 mice with MOG p35–55 to induce EAE. Systemic administration of mMSC led to an attenuation of disease severity as determined by the grade of paralysis. IDO−/−mMSC were equally effective in reducing disease severity as wild-type mMSC (Fig. 7A). We then used lymph node cells of immunized animals of all 3 groups and restimulated them in vitro with MOG p35–55. Lymphocytes from animals treated with wild-type or IDO1−/− mMSC showed reduced proliferation compared to lymphocytes from the control group. There was no difference between wild-type- and IDO1−/− mMSC-treated groups (Fig. 7B). While the production of the T-cell cytokines IL-2, IL-4, IL-6, IL-10, and IL-12/23p40 remained unchanged, treatment with both wild-type and IDO1−/− mMSC led to an equal reduction of IL-17 release from lymph node cells after restimulation with MOG p35–55 ex vivo. Interestingly TNF and IFN-γ seemed to be even more suppressed by IDO−/− mMSC than by the wild-type mMSC (Fig. 7C).
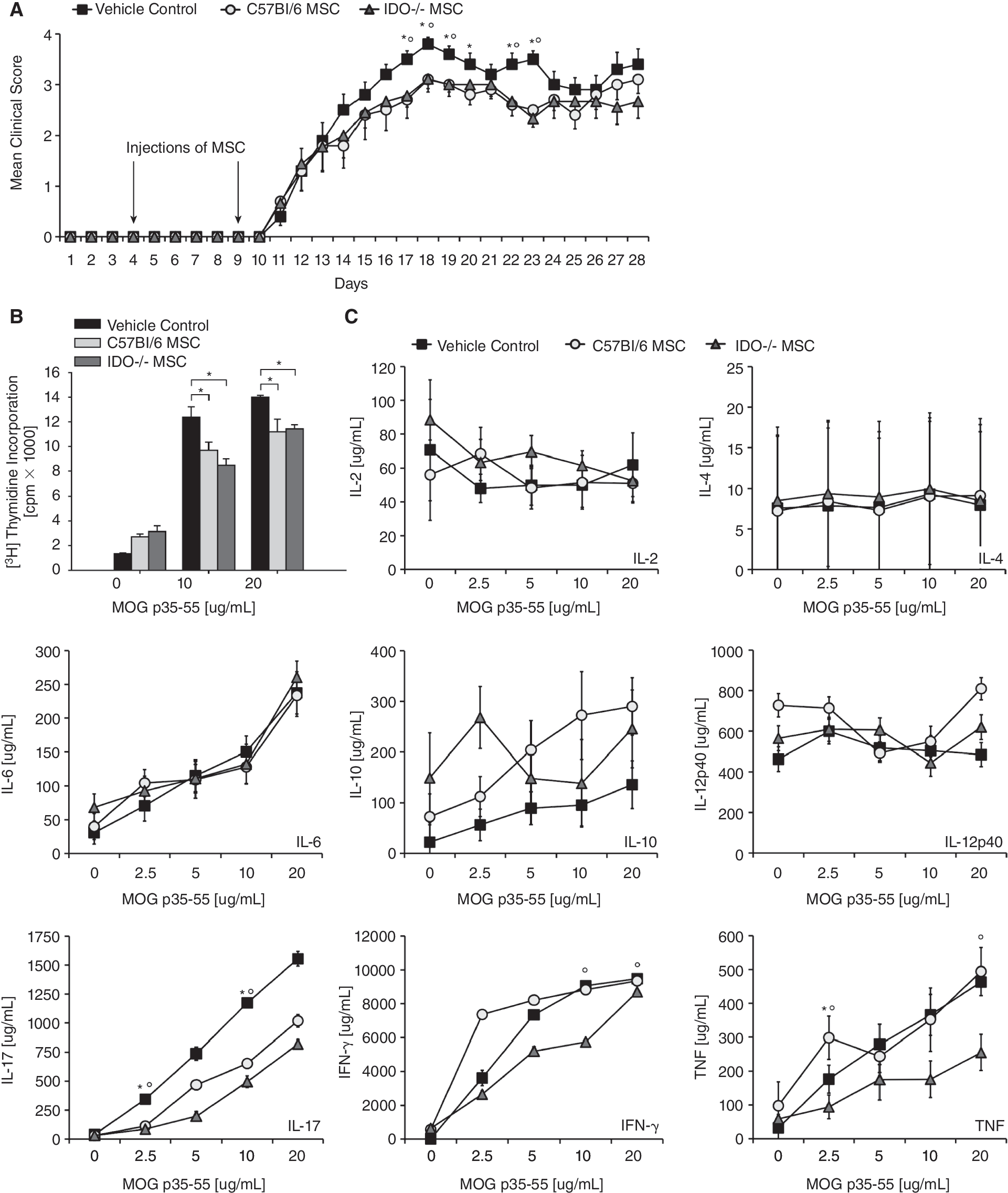
Indolamine-2,3-dioxygenase 1 (IDO1) is dispensable for mouse mesenchymal stem cells (mMSCs)-mediated immunosuppression in vivo. (
Discussion
MSC have the unique ability to suppress multiple cellular components of the immune system. The clinical development of hMSC as a source for tissue regeneration and as vehicles to deliver immunosuppressive signals to inflamed tissues after systemic administration has made rapid progress in the past several years. A recent phase II clinical trial has shown efficacy of systemic treatment of patients with steroid-resistant acute graft-versus-host disease with allogeneic bone marrow-derived hMSC [30]. Potential clinical applications have been extended to autoimmune diseases such as MS and rheumatoid arthritis, where preclinical mouse models have demonstrated the efficacy of autologous bone marrow-derived MSC [15]. The mechanism of immunosuppressive action of MSC has not been fully elucidated but appears to involve membrane-bound and soluble factors. Among the soluble factors, the oxidative catabolism of trp along the kyn pathway is an important immunosuppressive mechanism restricting antigen-specific T-cell responses in autoimmunity [24]. This pathway is operative in hMSC and delivers immunosuppressive signals to T cells, although it is dispensable when signaling through the IFN-g receptor is lacking [17]. We have recently shown that TLR ligands activate immunosuppressive trp catabolism via IDO1 in hMSC and that inhibition of IDO1 reverses hMSC-mediated T-cell paralysis [19]. This study was initiated to examine the contribution of IDO1-mediated immunosuppression to the therapeutic efficacy of mMSC in a mouse model of MS.
Using a transgenic mouse model with CD4+ T cells carrying a TCR specific for an immunodominant epitope of MOG, we show that autologous bone marrow-derived mMSC suppress proliferation of MOG-specific T cells (Fig. 5A) supporting the notion that suppression of encephalitogenic myelin-specific T cells is involved in the therapeutic efficacy of MSC in EAE [4].
We show, however, that as opposed to human MSC, mouse MSC cannot be activated to express functional IDO1 activity by stimulation with TLR ligands or with IFN-g. This inability to catabolize trp is likely to be species- and not strain-dependent, since neither C57Bl/6 MSC (Fig. 3B) nor phenotypically similar SJL/J MSC (Fig. 4B and 4C) displayed IDO1 enzyme activity (data not shown). This finding is in line with in vitro data from previous studies that failed to detect IFN-g-inducible IDO1 enzyme activity in murine MSC cell lines [31, 32]. Interestingly, IFN-g induced a strong IDO1 mRNA expression comparable to DC (Fig. 2) [TS: In artwork, Please Strike out A in Figure 2], pointing to post-transcriptional or post-translational mechanisms regulating IDO1 enzyme activity. An identical discrepancy between IDO1 mRNA expression and enzyme activity has been observed in CD8a+ DC [33]. The mechanisms involved in post-transcriptional and post-translational regulation of IDO1 enzyme activity remain elusive but may involve nitration of the enzyme by peroxynitrite [34].
As we and others measured IDO1 activity by determining the concentration of kyn in the cell culture supernatant via HPLC, it was conceivable that kyn produced from trp after IFN-g stimulation is catabolized further to downstream metabolites such as 3-hydroxykynurenine and 3-hydroxyanthranilic acid. The fact that trp levels in the supernatant of IFN-g-stimulated mMSC did not decrease and that the kyncatabolizing enzymes are down-regulated rather than up-regulated renders this hypothesis unlikely. Interestingly, stimulation with TLR ligands resulted in catabolism of kyn present in the cell culture media as evidenced by a reduction of kyn and simultaneous accumulation of a kyn catabolite in the supernatant (Fig. 3C and 3G). The kyn catabolite is unlikely due to activity of the kynurenine catabolizing enzyme KYNA as this enzyme is suppressed rather than induced by pI:C or LPS. KMO and 3-HAAO are not expressed by mMSC (Fig. 3D–3F). Moreover, the unidentified kyn catabolite (Fig. 3G) is not one of the classic catabolites such as kynurenic acid, 3-hydroxykynurenine, anthranilic acid, 3-hydroxyanthranilic acid, picolinic acid, quinolinic acid, kynuramic acid, or nicotinic acid as evidenced by HPLC analysis (data not shown). We are currently investigating the identity of this kyn catabolite.
The failure of the TLR ligands pI:C and LPS to induce functional trp catabolism led us to investigate whether TLR signaling in general is operational in bone marrow-derived mMSC. We show that mMSC express TLR1–9 mRNA at levels comparable to mDC, except for TLR7, TLR8, and TLR9 whose expression levels are 3 orders of magnitude lower than in mDC (Figs. 4A, 4C, and 6C). These results are in line with another study in mMSC [35]. As in human MSC [19], TLR3 and TLR4 are expressed at levels comparable to DC (Figs. 4A, 4C, and 6C). We show that the TLR pathway is not dysfunctional per se (Figs. 4D, 4E, 6D) indicating that—unlike in hMSC—activation of the TLR pathway alone is not sufficient to activate functional IDO1 in mMSC. It is tempting to speculate that IDO competence in mice is per se restricted to hematopoietic cells. Jasperson and colleagues have recently shown that IDO1-competent APC suppress GVHD, while IDO1 expression in epithelial cells was dispensable [36].
Our results identify an important immunological difference between human and murine MSC. In this respect, mMSC are similar to hDC where TLR activation requires a second signal to induce IDO1 [37]. It is tempting to speculate that the strong induction of an autocrine IFN-b loop by TLR ligation in hMSC [19] that is shared by plasmacytoid DC [38] is the critical determinant of the discrepancy between mMSC and hMSC.
Since these results did not rule out that IDO1 could be induced by other signals delivered through interacting T cells or myeloid cells, we tested whether inhibition of IDO1 would lead to an incapacitation of mMSC to suppress the activation of myelin-specific T cells. Treatment with the competitive IDO1 inhibitor 1-l-MT did not rescue the immunosuppressive phenotype in this setting (Fig. 5B). Interestingly, in the absence of mMSCs 1-l-MT suppressed T-cell proliferation not only indicating that IDO1-mediated trp catabolism does not restrict T-cell proliferation in this system but also suggesting that 1-l-MT may have off-target effects. It has been reported that 1-l-MT modulates the function of APC independently of IDO1 inhibition [39]. To more specifically delineate the role of IDO1 in immunosuppression of mMSC, we generated MSC from bone marrow cells of IDO1-deficient mice. IDO1−/− MSC were phenotypically similar to wild-type MSC with respect to surface marker expression, differentiation, expression of TLR, and IL-6 induction (Fig. 6A–6D). IDO1 deficiency in mMSC neither reversed mMSC-mediated suppression of antigen-specific T-cell proliferation in vitro (Fig. 6E) nor did it alter the therapeutic effects of mMSC in a mouse model of MS (Fig. 7A). Interestingly, both wild-type and IDO1−/− MSC led to a reduction of IL-17 production in myelin-specific CD4+ T cells ex vivo while IFN-g and TNF release was even more suppressed by IDO-deficient mMSC (Fig. 7C). Rafei and colleagues have recently demonstrated a suppression of both TH1 and TH17 immunity by MSC [9, 40]. The indistinguishable clinical outcome and the lack of differences in TH cell proliferation between the 2 groups may indicate that mMSC are responsible for TH17 suppression rather than TH1 polarization of myelin-specific T cells in vivo. TH17 cells have shown to be critically involved in the pathogenesis of EAE, although their ultimate importance in EAE and in other models of autoimmune diseases remains controversial [41].
Conclusion
We show that IDO1 is dispensable for the immunosuppressive signals delivered by mMSC and for the therapeutic effects of autologous mMSC in autoimmune neuroinflammation. This is in sharp contrast to human MSC where IDO1 is critically involved in the suppression of T-cell immunity [16, 19]. The preclinical development of MSC as a cell-based therapy for autoimmune diseases in mouse models using autologous bone marrow-derived MSC should therefore be interpreted with caution with respect to its prediction for the therapeutic efficacy of hMSC in human autoimmune diseases. Using hMSC in mouse models, which can effectively be done without obvious evidence of immune rejection [42], may be more appropriate for preclinical trials characterizing and optimizing route of administration, motility, cellular targets, and immunosuppressive mechanisms of MSC in disease settings.
Footnotes
Acknowledgments
This work was supported by the Hertie Foundation and grants from the Center for Interdisciplinary Research Tübingen (1496–0-0 to MP and 1546–0-0 to W.W. and M.P.), by the Helmholtz-Foundation (VH-NG-306) to M.P., by the Biomedical Sciences Exchange Program (BMEP) to T.V.L. We thank Sabrina Koch and Andreas Mlitzko from Heidelberg-Pharma for expert technical assistance. We thank Dr. Uta Opitz for help with the HPLC measurements and interpretation of these data.
Author Disclosure Statement
No competing financial interests exist.