
Abstract
One of the challenges in stem cell research is to avoid transformation during cultivation. We studied high passage subventricular zone derived neural stem cells (NSCs) cultures of adult rats in the absence of growth factors epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF). We termed this culture exogenous growth factor independent neural stem cells (GiNSCs). GiNSCs expressed stemness markers, displayed a high constitutive NF-κB activity and an increased, aberrant, polyploid DNA content. GiNSCs showed a tumorigenic phenotype and formed colonies in a soft agar assay. Microarray analysis showed the up-regulation of the NF-κB target gene vascular endothelial growth factor (VEGF). In contrast, proneuronal genes were down-regulated. Under neuronal differentiation conditions GiNSCs adopted a glioma-like phenotype, with nuclear p53, preserving high amounts of Nestin positive cells and prolonged proliferation. Neutralization of VEGF strongly inhibited proliferation and induced differentiation. In a gain of function approach, the transfection of NSCs with constitutively active upstream kinase IKK-2 led to constitutively activated NF-κB, proliferation in absence of growth factors and augmented VEGF secretion.
In a rescue experiment a reduction of NF-κB activity by overexpression of IκB-AA1 was able to shift the morphology toward an elongated cell form, increased cell death, and decreased proliferation. Thus GiNSCs may provide a potent tool in cancer research, as their exogenous cytokine independent proliferation and their constitutively high NF-κB expression presumes cancerous properties observed in gliomas. In addition, this study might add a novel mechanism for detecting oncogenic transformation in therapeutic stem cell cultures.
Introduction
S
Such NSCs have been isolated from several regions of the mammalian brain, including the dentate gyrus [1], subcortical white matter [2], and the subventricular zone (SVZ) with the latter one considered being the largest of these germinal centers [3].
Characterization of NSCs in vitro is assessed by the expression of the intermediate filament Nestin [4], the RNA-binding Protein Musashi-1 [3,5], and the transcription factors Sox1 [6] and Sox2 [7].
Many experimental studies have implicated the NSCs residing in the SVZ as a source of oncogenicity and tumor formation [8,9] in vivo. In particular there is compelling evidence that gliomas arise from NSCs that have undergone malignant transformation [10].
Cell proliferation and cell numbers within the developing and adult CNS are tightly controlled under physiological conditions [11,12].
In a previous study, we were able to show that tumor necrosis factor (TNF) not only triggers the aggregation of 3D neurophere NSC cultures but also the proliferation of NSCs in vitro [13,14]. The latter one proved the known transduction of TNF to the IKK-α/β-complex [15] to apply to NSCs as well. In both approaches we found TNF signaling via the NF-κB pathway to have huge impacts.
NF-κB proteins in mammals are dimers assembled from a set of at least five different subunits termed p50, p52, p65/RelA, RelB, and c-Rel with p65/p50 dimers being the most frequent ones within the CNS [16]. Normally, NF-κB proteins are retained in the cytoplasm by a group of inhibitory proteins known as IκBs keeping them in an inactive state [17]. This trimeric NFκB–IκB complex can be activated by various stimuli such as cytokines, neurotransmitters, mitogens, and growth factors [17,18].
NF-κB transcription factors are crucially important in a wide variety of biological processes such as immune and inflammatory responses [19], neuroprotection and degeneration [17], and pathological tumor malignancies [15,20]. Inflammatory responses in turn are a prerequisite for immune defense and angiogenetic processes. As cancer frequently is associated with maladaptive chronic inflammation, it is interesting to note that TNF is described to strongly activate NF-κB in a row of experimental cancer models [15]. Hence, the role of NF-κB in cancer development and progression has been discussed very extensively [20 –23].
NSCs are hoped to be capable of replacing lost cells within the CNS because of their therapeutic potential in neurodegenerative disorders [24,25] and cancers [26]. Yet, as one of the crucial features of NSCs is the ability to proliferate, they eventually can accumulate genetic aberrations leading to tumorigenesis.
Materials and Methods
Isolation and culture of adult NSCs
Tissue was extracted from Wistar rats according to local guidelines (Bezirksregierung Detmold). NSCs from the lateral SVZ of 3–5 adult rats/preparation were isolated and cultivated as described in reference [13].
Isolation of GiNSCs
Subcultured secondary NSCs, primordially isolated from the SVZ of adult rats, were maintained in culture for 92 passages. After dissociation with Accutase cells were seeded on poly-
Semiquantitative RT-PCR
A total of 4.0 × 106 cells of NSCs or GiNSCs, 50 μg of cortex or liver tissue were harvested per sample and total RNA was isolated with RNeasy (Qiagen, Valencia, CA) according to the manufacturer's instructions. cDNA was subsequently synthesized using SuperScript III First-Strand Synthesis System for reverse transcription-polymerase chain reaction (RT-PCR; Invitrogen, Carlsbad, CA) following manufacturer's guidelines.
Microarrays
Microarray analysis, data processing and handling were performed as described by Schwamborn et al.[27].
Immunocytochemistry
Neurospheres were harvested, fixed, and stained as described in reference [13]. The following antibodies were used: Nestin 1:100 (Milipore, Billerica, MA), Musashi 1:100 (Chemicon, Temecula, CA), Sox2 1:150 (Chemicon), Notch2 1:100 (Santa Cruz, Santa Cruz, CA), PSA-NCAM 1:100 (Chemicon) GFAP 1:100 (Chemicon), β-III-tubulin 1:100 (Promega, Madison, WI), Gad67 1:100 (Chemicon), Neurofilament 1:100 (Chemicon), PCNA 1:100 (Chemicon), p53 1:100 (Santa Cruz), and p65 1:100 (Chemicon).
Proliferation assays
The determination of proliferation of NSCs and GiNSCs spheres was performed as described in reference [13]. NSCs and GiNSCs conducted to neuronal or glial differentiation were seeded in an initial density of 1.0 × 105 cells and cell numbers were determined by a trypan blue exclusion assay daily.
Alternatively, NSCs and GiNSCs induced to neuronal or glial differentiation for 5 or 4 days, respectively, were incubated with a bromodeoxyuridine (BrdU) solution of the BrdU Labeling and Detection Kit (Boehringer Mannheim, Mannheim, Germany) for 2 h, fixed and stained for BrdU incorporation according to the manufacturer's instructions. Differences in proliferation between NSC and GiNSC were assessed by 2-way analysis of variance (ANOVA) followed by post hoc t-test with Bonferroni correction. P ≤ 0.05 was considered as significant.
Cell differentiation
To analyze the neuronal and glial differentiation potential of NSCs and GiNSCs, cells were differentiated as described earlier [13].
Differences in marker expressions between differentiated NSC and GiNSC were determined by 2-way ANOVA followed by post hoc t-test with Bonferroni correction. P ≤ 0.05 was considered significant.
Reporter gene assay and rescue approach (transfection and analysis)
Neurospheres were dissociated as described above and transfected using a Rat NSC Nucleofector Kit (Lonza, Cologne). To detect NF-κB activity in NSCs, a Dual-Luciferase Reporter Assay System (Promega, Madison, WI) and κB-luc Reporter (BD Clonetech, Mountain View, CA) were used as described in reference [13]. In addition the IκB-AA1 [28] or caIKK2 (a constitutively active IKK2), cloned into the pLenti4/V5-DEST expression vector (Invitrogen, Carlsbad, CA) were cotransfected. A representative experiment is shown.
For a phenotype rescue approach GiNSCs were conducted to neuronal differentiation. Upon differentiation, the cells were either transfected with 2 μg of the IκB-AA1 or the mock vector pMETalpha. All samples were additionally transfected with 1 μg of the pmaxGFP (Lonza). After 5 days of growth, the cultures were processed for immunocy-tochemistry and proliferation assays. Differences in marker expressions and proliferation between NSC and GiNSC were determined by 2-way ANOVA followed by post hoc t-test with Bonferroni correction. P ≤ 0.05 was considered significant.
Cell cycle analysis using FACS
Nuclei were isolated by hypotonic lysis. 5 × 105 cells were harvested and plasma membranes lysed for 30 min by a hypotonic lysis solution (0.1 % w/v Sodium citrate, 0.1% v/v Triton X-100). Nuclei were stained with 0.1% v/v propidium iodide and cell aggregates were removed by filtration through a 40 μm mesh. Nuclear staining was analyzed on a FACSCalibur flow cytometer (Becton Dickinson, Germany) and data processed with the WinMDI 2.8 software (J. Trotter, USA). Differences in DNA content between control NSCs and GiNSCs were assessed by 2-way ANOVA followed by post hoc t-test with Bonferroni correction. P ≤ 0.05 was considered significant.
Preparation of rat metaphase chromosomes
A number of each 1 × 107 cells of NSCs and GiNSCs grown in tissue flasks were incubated for 2 h at 37°C in medium containing colcemide (0.05 μm/mL) to arrest cells in mitotic metaphase. After treatment with prewarmed hypotonic solution (0.075M KCl) for 15 min at 37°C the cells were progressively fixed three times in methanol/acetate (3:1). The suspension was dropped from a height of 40 cm onto cleaned wet coverslips and air dried. Chromosomes were stained with DAPI (50 ng/mL) and mounted with moviol (Hoechst).
Western blot analysis
GiNSCs, NSCs, and NSCs stably transfected with caIKK2 were cultured for 48 h (for transfected NSCs) or 72 h without media change. After centrifugation at 200g the supernatants (3 mL) were collected and concentrated to 100 μL using Amicon 10 K centrifuge spin column (Millipore, Temecula, CA). The blotting was performed according to Widera et al. [13] using polyclonal antibody against vascular endothelial growth factor (VEGF; R&D Systems, 0.2 μg/mL) or FLAG-tag (Sigma-Aldrich, St. Louis, MO; 1:5000) with gentle agitation.
Neutralization of VEGF and determination of proliferation
GiNSCs were cultivated as described above with supplementation of IgG-control or 0.1,0.5, and 1 μg/mL of neutralizing α-rat VEGF antibody (R&D Systems, Minneapolis, MN). For BrdU incorporation analysis 10 μM BrdU was added every 24 h. BrdU incorporation was analyzed by immunocytochemistry. Antibody staining was visualized using confocal laser scanning microscopy (LSM 510l; Zeiss, Germany) and analyzed using ZEN software (Zeiss, Germany).
Measurement of tumorigenicity using soft agar assay
Anchorage independent cell proliferation was investigated by a soft agar assay. Top agarose was prepared from Agarose (Biozym) in Dulbecco's modified Eagle's medium (DMEM)/F12 substituted with B27 supplement. Cells were seeded at a density of 3 × 103 cells per cell culture well in 0.35% top agarose and cultured for 14 days on 0.5% base agar (Difco Agar Noble; Becton Dickinson, Franklin Lakes, NJ) at 37°C. GiNSCs and NSCs were cultured in absence of EGF and bFGF. As control NSCs with EGF/bFGF were used. The medium was changed every 3–4 days. When present, EGF and bFGF was added to both the soft agar and the medium at 20 ng/mL each. After 14 days of culture, cells were stained with 0.05% crystal violet for 2 h at 4°C. Colonies were counted in the entire well per condition under a dissecting microscope.
Generation of GiNSCs via stable transfection with caIKK2
NSCs (passage 14) were transfected using Amaxa (see above) with vector carrying caIKK2 [29], puromycin resistance and FLAG-tag or with a vector containing puromycin resistance and FLAG-tag alone as control. Transfected cells were cultured in medium with EGF and bFGF for 24 h followed by treatment with 1.5 μg/mL puromycin. Puromycin containing medium was changed every 24 h. After 72 h of culture in presence of puromycin, the medium was replaced by standard EGF/bFGF containing medium followed by culture for 72 h at 37°C. Thereafter the growth factors were deprived and the cells (1 × 106/condition) were cultivated for additional 48 h followed by analysis of cell morphology, cell number, western blot or transfection for reporter gene assay (see above). In a control approach EGF and bFGF were left in the medium. In all approaches successful transfection was verified via western blot against the FLAG-tag.
Results
Characterization of exogenous G rowth factor i ndependent N eural S tem C ells (GiNSCs)
SVZ derived NSCs were kept as in vitro culture for 92 passages. These NSCs were exposed to differentiation promoting conditions in the absence of EGF and bFGF. We noticed a few cells remaining in suspension. Interestingly, these cells did not adhere to the substrate as the majority of cells and they appeared vital. To analyze this population of differentiation nonresponding cells, we harvested them and kept them in culture in the absence of growth factors (Fig. 1A). To our surprise, the cells were vital and, moreover, within 14 days began to proliferate and to form neurospheres. As these cells were able to form neurosphere like clusters in the absence of EGF/bFGF supplementation we termed them GiNSCs. GiNSCs were cultured for at least 20 passages.
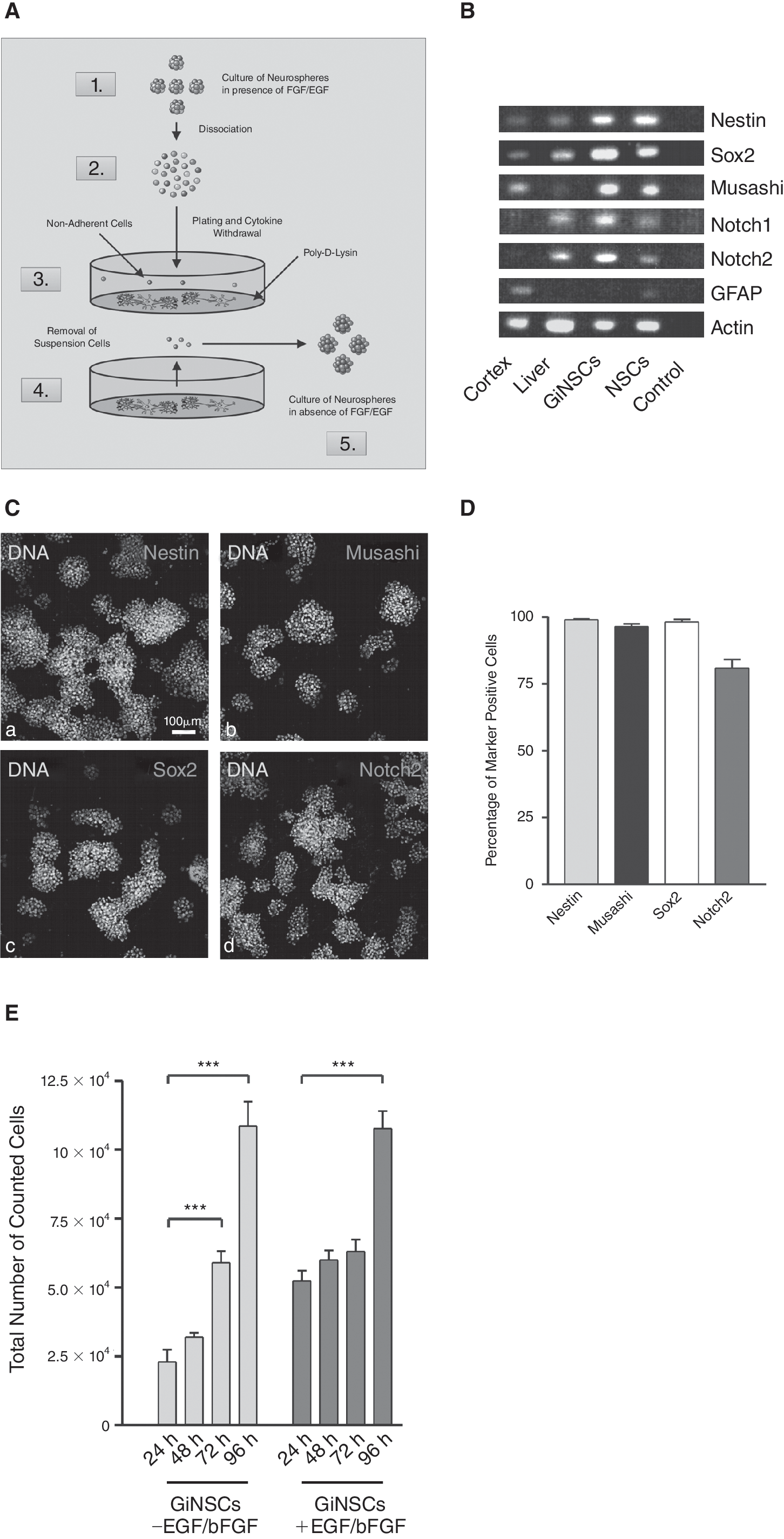
A neural stem cell (NSC) population independent of growth factor supplementation (GiNSCs). (
Expression pattern of GiNSCs was studied by semiquantitative RT-PCR (Fig. 1B). NSC markers Sox-2, Musashi, Nestin [3], and Notch [30] were amplified. To verify PCR-analysis immunocytochemistry was performed (Fig. 1C). Most of the GiNSCs were Nestin immunoreactive, and featured high levels of Musashi, essential for neurosphere formation and proliferation [3]. The NSC-specific transcription factor SOX2 [31] and Notch2, a marker present in proliferating neural cells [30] were detected as well (Fig. 1C and 1D). Moreover, we found expression of PSA-NCAM, a marker for migrating neuronal precursor cells. The differentiation markers GFAP and β-III-tubulin (data not shown) were not expressed (Fig. 1C).
GiNSCs proliferate without growth factors
To analyze, if a growth factor supplementation with EGF/bFGF still had an effect on proliferation, samples in presence or absence of growth factors were prepared (Fig. 1E). Initial cell number of 1 × 104 increased to over 1 × 105 within 96 h (average doubling time of 28.9 h). In comparison, cultivation of GiNSCs in presence of EGF and bFGF, though yielding a more considerable increase during the first 24 h, did not lead to a higher total cell number after 96 h. Without EGF and bFGF addition population doubling time within the first 24 h was 18.16 h.
GiNSCs display a glioma phenotype upon differentiation
In neuronal differentiation attempts morphology of GiNSCs differed notable from that of the control NSCs (Fig. 2A). After 5 days GiNSCs appeared compact, with glial-cell like protrusions positive for Nestin, in contrast to the typical neuronal morphology. Moreover, expression patterns differed significantly between control NSCs and GiNSCs, as the latter ones were highly positive for Nestin (Fig. 2B). Moreover, GiNSCs displayed a higher percentage of GFAP positive cells and strikingly less numbers of β-III-tubulin expressing cells. Although NF-M [32,33] expression was at similar levels, GiNSCs had slightly less GAD67 [34] positive cells than NSCs. In addition, a tremendous morphological difference to NSC cells was apparent (Fig. 2A, a–i).
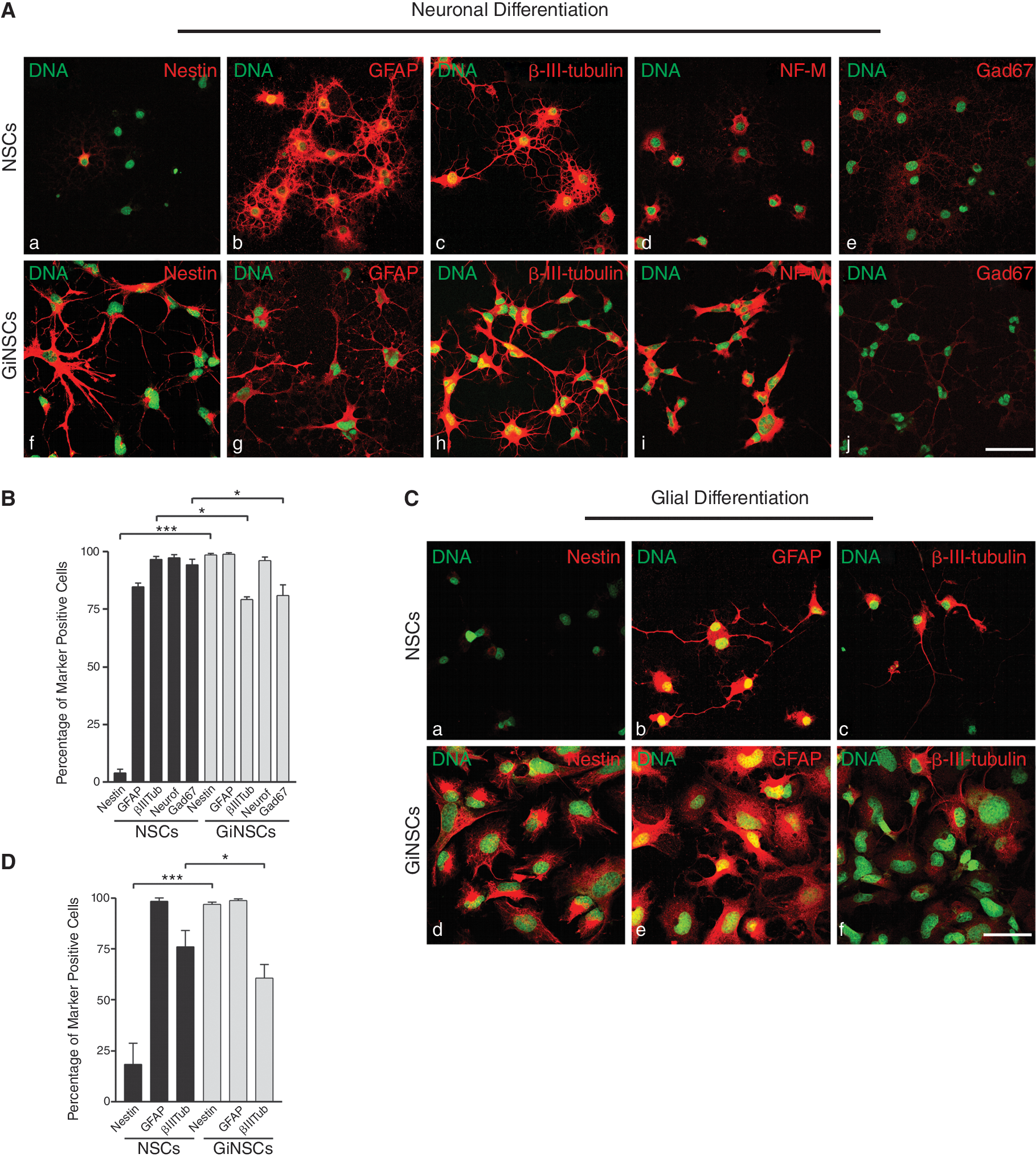
Growth factor independent neural stem cells (GiNSCs) feature an altered phenotype when prompted to differentiate. (
Conducted to glial differentiation GiNSCs appeared to be very compact in shape and possessed enlarged nuclei (Fig. 2C). The glial like phenotype differed from the appearance of the NSCs controls. GiNSCs displayed the same drift in Nestin expression as they did in the neuronal differentiation (Fig. 2D).
GiNSCs continue proliferating upon differentiation
During neuronal and glial differentiations we observed the cell density of GiNSCs to increasingly shift in comparison to NSC controls. As cells for all samples were initially seeded in the same density and this phenomenon appeared in all independent differentiation approaches we assumed such a variation to be due to proliferation of GiNSCs.
After 5 days of neuronal differentiation cells were incubated with BrdU, followed by immunocytochemical detection of BrdU incorporation (Fig. 3A). While NSCs controls had a rather discrete to no presence of BrdU, nuclei of GiNSCs showed to take up marked amounts of BrdU. Quantification proved nearly 70% of GiNSCs to be highly BrdU positive, which was in significant contrast to <5% BrdU positive NSCs (Fig. 3A). In contribution a trypan-blue exclusion assay displayed that GiNSC cell numbers doubled within 72 h whereas NSC numbers remained invariable (Fig. 3C).

Differentiated growth factor independent neural stem cells (GiNSCs) continue to proliferate. (
Analogous to neuronal differentiation, GiNSCs induced to glial differentiation incorporated BrdU as well (Fig. 3B). Similarly a trypan-blue exclusion assay revealed a tripled GiNSC cell number within 72 h, hence being significantly more cells compared to NSCs (Fig. 3D).
GiNSCs form colonies in soft agar assay
Anchorage independent proliferation was tested using soft agar assay (Fig. 4). After 14 days of culture GiNSCs formed 214 ± 37 colonies in medium without EGF/bFGF. In contrast NSCs control cultures in presence of growth factors formed no colonies at all, although single cells remained viable and showed signs of sprouting (see Fig. 4). In a control approach with NSCs cultivated without EGF and bFGF no viable cells could be detected.

Measurement of tumorigenicity using soft agar assay. Cells were seeded at a density of 3 × 103 cells per cell culture well in 0.35% soft agar and cultivated for 14 days at 37°C. Growth factor independent neural stem cells (GiNSCs) and neural stem cells (NSCs) were cultured in absence of epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF). As control NSCs with EGF/bFGF were used. If present, EGF and bFGF were added to both the soft agar and the medium at 20 ng/mL each. After staining with crystal violet, the number of colonies has been determined. GiNSCs formed 214+ colonies, whereas NSCs with growth factors show no colony formation but sprouting of single cells (see blow-up in the middle panel). In a control approach using NSCs without growth factors, no viable cells have been detected.
GiNSCs accumulate nuclear p53 and have a constitutively high NF-κB activity
A very common feature among tumor cell lines as well as tissues obtained from primary cancers is the nuclear accumulation of the tumor suppressor protein p53 accompanied or caused by its loss of function. For this reason we analyzed GiNSCs for nuclear accumulation of p53 during neuronal differentiation (Fig. 5A). In GiNSCs p53 displayed huge nuclear accumulation, whereas the NSC control had rather low amounts of p53 (Fig. 5A, a and b), predominantly located in the perinuclear space, as assessed by nuclear counterstaining (Fig. 5A, c–f). Nuclear p53 was found in almost 45% of all GiNSC cells, contrasted by <5% in NSC controls (Fig. 5B).
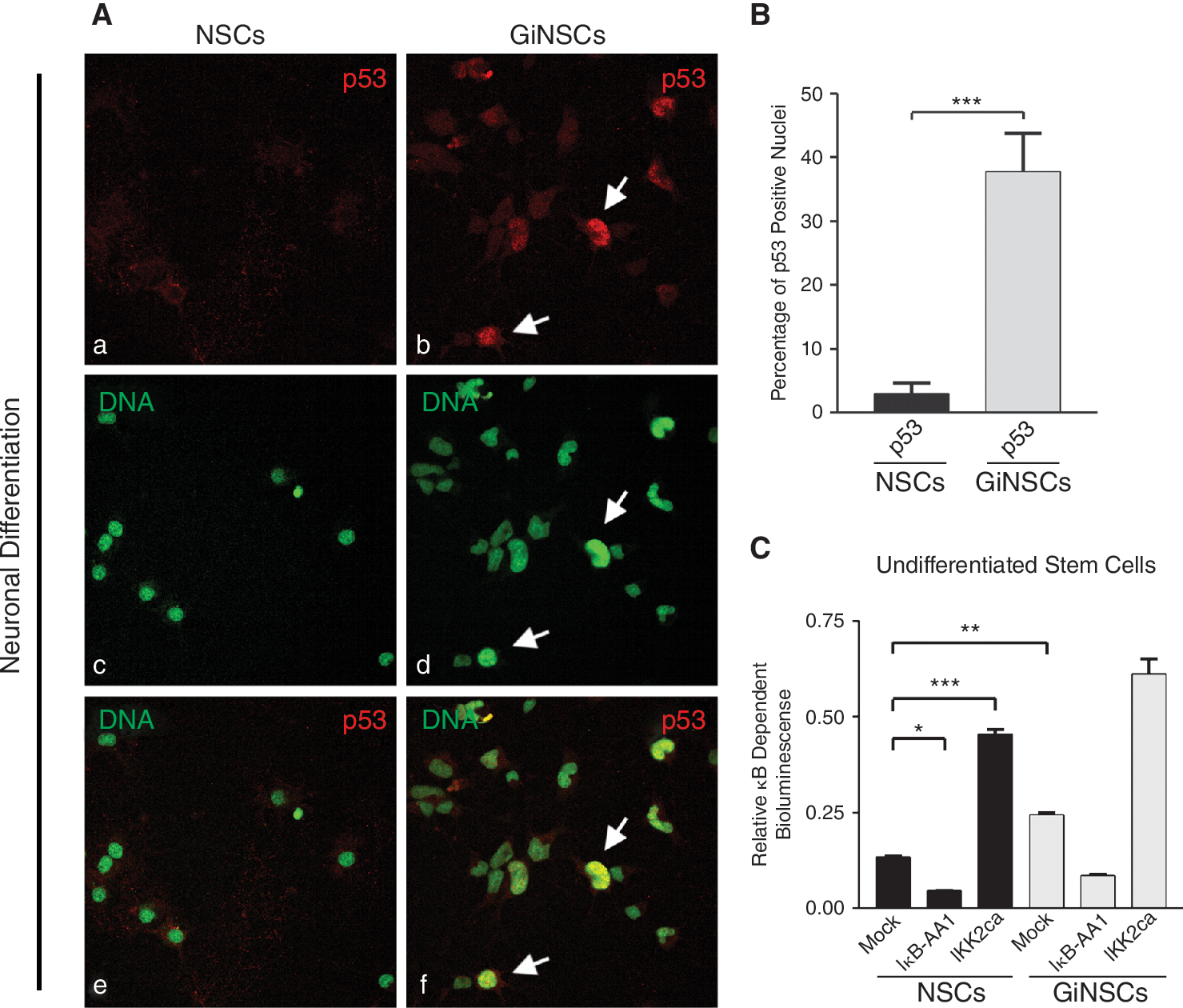
Growth factor independent neural stem cells (GiNSCs) accumulate nuclear p53 and exert a high constitutive NF-κB activity. (
When eliciting regulation downstream of p53, it is noteworthy, that activated p53 was reported to lead to activated NF-κB in Saos-2 tumor cells [35]. Therefore, we wondered whether the independence of GiNSCs from growth factor supplementation might correlate with an altered NF-κB expression.
Investigation of NF-κB signal transduction was conducted by reporter gene analysis. Cotransfection of the NF-κB reporter plasmid and IκB-AA1 significantly decreased the activity measured, whereas the constitutively NF-κB activating caIKK2 significantly increased the κB-dependent bioluminescence. Notably, basal NF-κB activity in mock-transfected control cultures was significantly higher in GiNSCs than in mock transfected NSCs, indicating a high amount of constitutive NF-κB activity in GiNSCs.
GiNSCs harbor an aberrant DNA content
We analyzed the cell cycle of GiNSCs via FACS to quantify major differences to NSCs in the degree of the polyploidy. Normally, a stem cell population such as NSCs displays 3 subpopulations (Fig. 6A): A peak of cells with a single chromosome set (1n) that either are arrested in G0 state or have undergone cytokinesis currently being in replication state (S-phase) and another peak of cells that already have doubled their DNA (G2/M). In contrast to this GiNSCs additionally had a dispersed population of polyploid cells. In detail GiNSCs had a significantly higher percentage of cells being in G2/M phase and a significant number of cells possessing more than 2 chromosome sets (polyploidy, Fig. 6B).
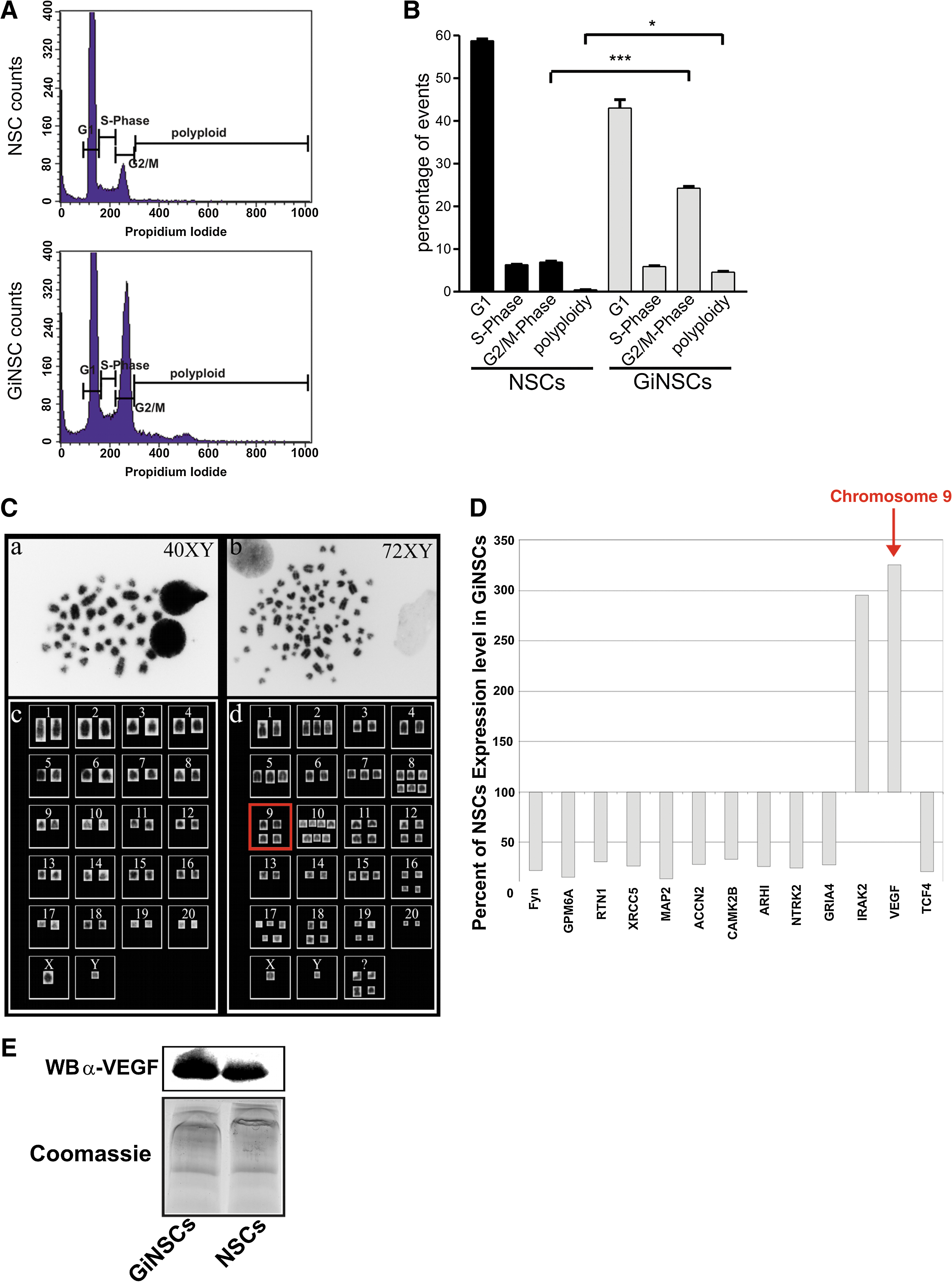
(
To further analyze to what extend the DNA amount of GiNSCs had grown, we investigated their chromosome sets via karyotyping metaphases (Fig. 6C). Inverted DAPI-stained images of a NSCs metaphase (Fig. 6C, a) showed the expected 20 chromosome pairs plus 2 gonadosomes, typically of rat cells. Quite strikingly GiNSC metaphases (Fig. 6C, b) verified that GiNSCs harbor a dramatically increased chromosome number (Fig. 6C, a and b): 72 chromosomes and 2 gonadosomes, many of them constituting for more then 2 individual chromosomes each pair, and 4 pleiomorphic chromosomes that were not sortable to any of the 20 chromosome types (Fig. 6C, c and d).
Microarray analysis showed upregulation of VEGF and IRAK2
We visualized shifts in gene-expression patterns by microarray analysis. In particular, we were interested whether multiplication of chromosome pairs could be correlated with multiplications in gene expression.
Microarrays were processed with total RNA of GiNSCs and control NSCs. Targets, involved in a range of cellular tasks such as cytoskeleton assembly, neuronal differentiation and tumor suppression were selected with regard to their corresponding chromosomes, shown to be amplified in GiNSCs. Among those the angiogenesis factor VEGF [36] from chromosome 9 (9q12), with 4 individual chromosomes in GiNSC karyotyping (Fig. 6C, d), was increased in GiNSCs 3-fold. Interestingly VEGF is a NF-κB target gene [37]. Similarly, the downregulation of notch 1 inhibited cancer cell invasion by decreasing NF-κB DNA binding activity and subsequent VEGF downregulation [38]. IRAK2, involved in NF-κB dependent survival signaling [39], though its corresponding chromosome 4 was not found to be amplified, had 3 times higher expression levels than NSCs as well. Other targets, such as the DNA repair protein XRCC5 (9q34) [40], the neural cytoskeleton component MAP2 (9q32) [41] or the cation channel subunit ACCN2 (7q36) [42] were drastically downregulated (Fig. 6D). Candidate gene expression values of NSCs were set as 100% with GiNSC values set in relation to this relative baseline. A number of neuronal genes such as CamK2B, MAP2, ACCN2, and Fyn were found to be downregulated. Since patient derived gliomas rely on VEGF for growth we focused on the analysis of VEGF protein expression. Hence, we screened the VEGF expression in supernatants from cultured GiNSCs in comparison to control NSCs. Western blotting revealed a high amount of secreted VEGF in supernatants prepared from GiNSC and corroborated the microarray results (Fig. 6E).
Stable transfection of NSCs with caIKK2 leads to GiNSC-like phenotype in NSCs
In order to investigate the potential transformation of NSCs by constitutively activated NF-κB, NSCs (passage 14) were stably transfected with a constitutively active form of IKK-2 (caIKK2) carrying vector with additional puromycin resistance and FLAG-tag or puromycin resistance and FLAG-tag alone (Fig. 7A). IKK-2 is the upstream kinase activating NF-κB. The success of the transfection has been verified via western blot against FLAG-tag (data not shown). After 48 h of culture in absence of growth factors no cell proliferation has been observed in mock-transfected cells (Fig. 7C). In addition the morphology of the cells changed to more differentiated phenotype than caIKK2-transfected cells (Fig. 7B). In contrast, caIKK2-transfected cells proliferated moderately after growth factor deprivation. Furthermore, an increased NF-κB activity has been observed in caIKK2 transfected cells compared to mock transfected cells in presence or absence of growth factors (Fig. 7D). Finally, NSCs stably transfected with caIKK2 showed elevated level of the secreted NF-κB target gene VEGF compared to mock-transfected NSCs (Fig. 7E). Taken together, a constitutive activation of NF-κB using caIKK2 led to a GiNSC-like phenotype in NSCs.
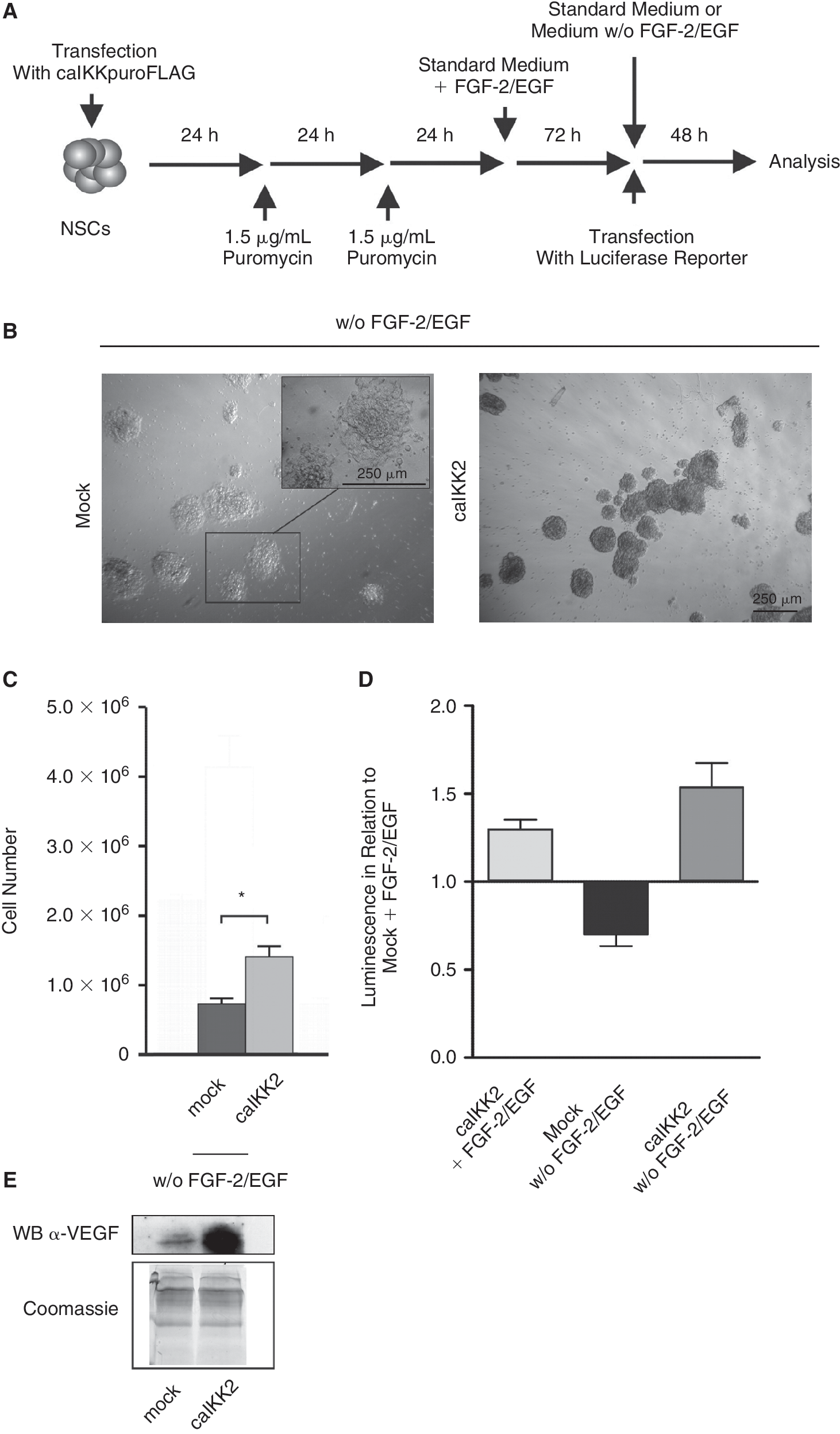
Stable transfection of neural stem cells (NSCs) with caIKK2 leads to growth factor independent neural stem cell (GiNSC)-like phenotype in NSCs. (
Neutralization of VEGF results in decreased proliferation of GiNSCs
Cultures of GiNSCs were treated with a neutralizing antibody directed against rat VEGF for 48 and 72 h (Fig. 8A). Differences in neurosphere size and number were evident after 48 h and in addition after 72 h some stem cells started to differentiate (Fig. 8A). Control IgG did not interfere with proliferation (upper panel in Fig. 8A). Cell proliferation decreased in GiNSCs treated with anti-VEGF antibody (Fig. 8B). High power magnification showed nuclear BrdU incorporation under control conditions, which was nearly invisible after anti-VEGF treatment.
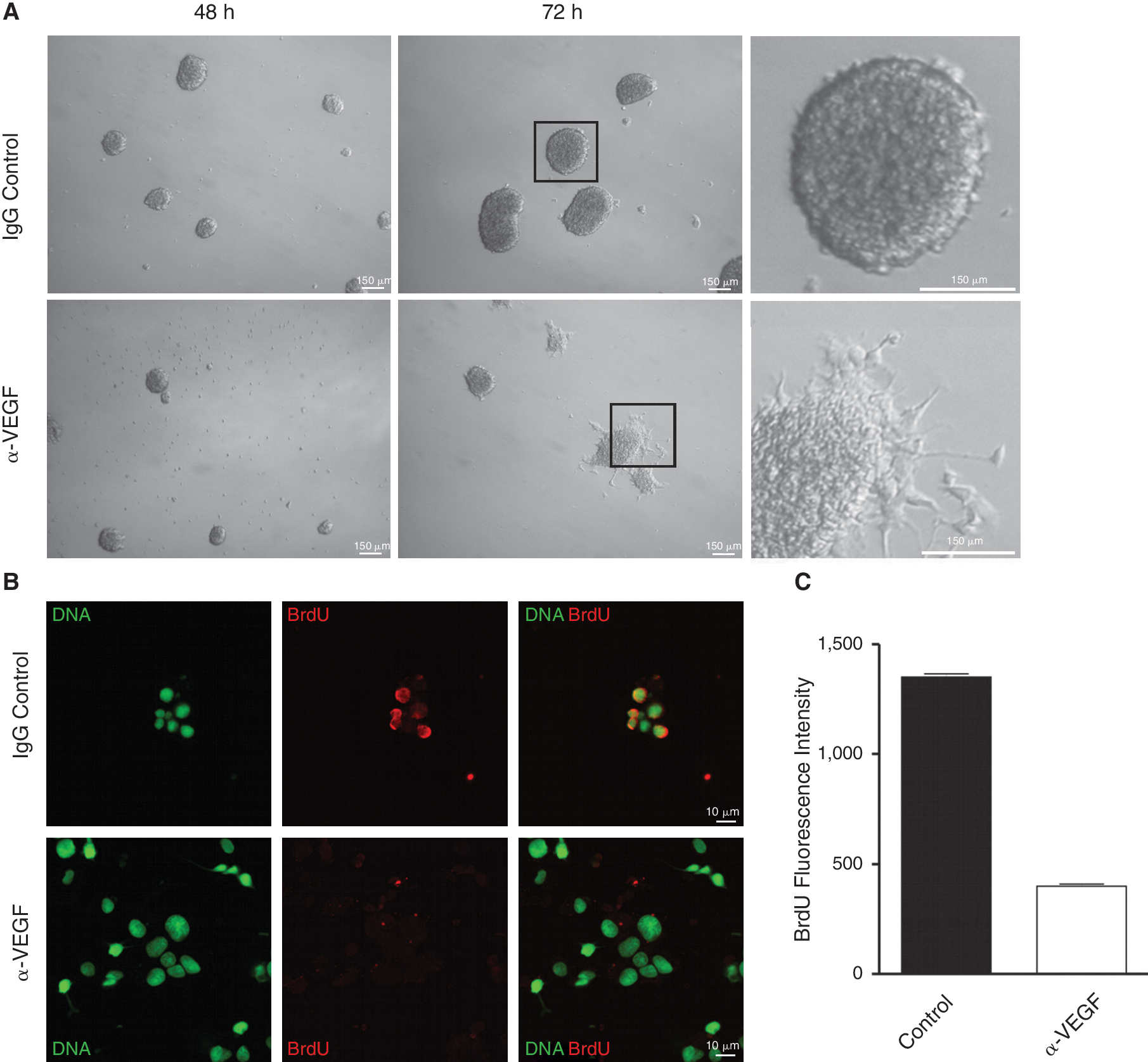
Neutralization of vascular endothelial growth factor (VEGF) leads to decreased proliferation and induces differentiation of growth factor independent neural stem cells (GiNSCs). (
Reduced NF-κB activity reverses the tumorigenic phenotype in differentiating GiNSCs
We hypothesized that a reduction of NF-κB activity in GiNSCs might reduce the proliferation rate and induces cell death.
As a sort of rescue approach we introduced IκB-AA1 into GiNSCs previous to induction of neuronal differentiation. An additional approach with a mock vector served as control. 5 days after adhesion of the cells to the provided substrate marker expression was analyzed. Mock transfected GiNSCs had the same compacted cell shape with glial-cell like protrusions as previously observed among untransfected GiNSCs (Fig. 9A, a–c). In contrast IκB-AA1 transfected GiNSCs differed remarkably and instead had an elongated shape (Fig. 9A, d–f). Differentiated cells were screened for their expression profiles of Nestin, GFAP, and β-III-tubulin (Fig. 9A, a3–f3). To assess the influence of NF-κB activity GFP reporter positive cells were analyzed exclusively (Fig. 9A, a2–f2). Approximately 55% of the GFP positive cells of the IκB-AA1 transfected approaches showed an elongated phenotype compared to <10% in Mock transfected approaches (data not shown). IκB-AA1 transfected cells still were positive for Nestin, but to a lesser percentage (Fig. 9B). Quantifications of GFAP showed a reduction of positive cells and a slight increase of neuronal marker β-III-tubulin in IκB-AA1 transfected GiNSCs. Immunodetection of activated NF-κB subunit p65 verified that IκB-AA1 transfected GiNSCs had less activated p65 (Fig. 9C). Quantification of p65 immunodetection revealed IκB-AA1 transfection to half the amount of p65/GFP double positive nuclei (Fig. 9D).
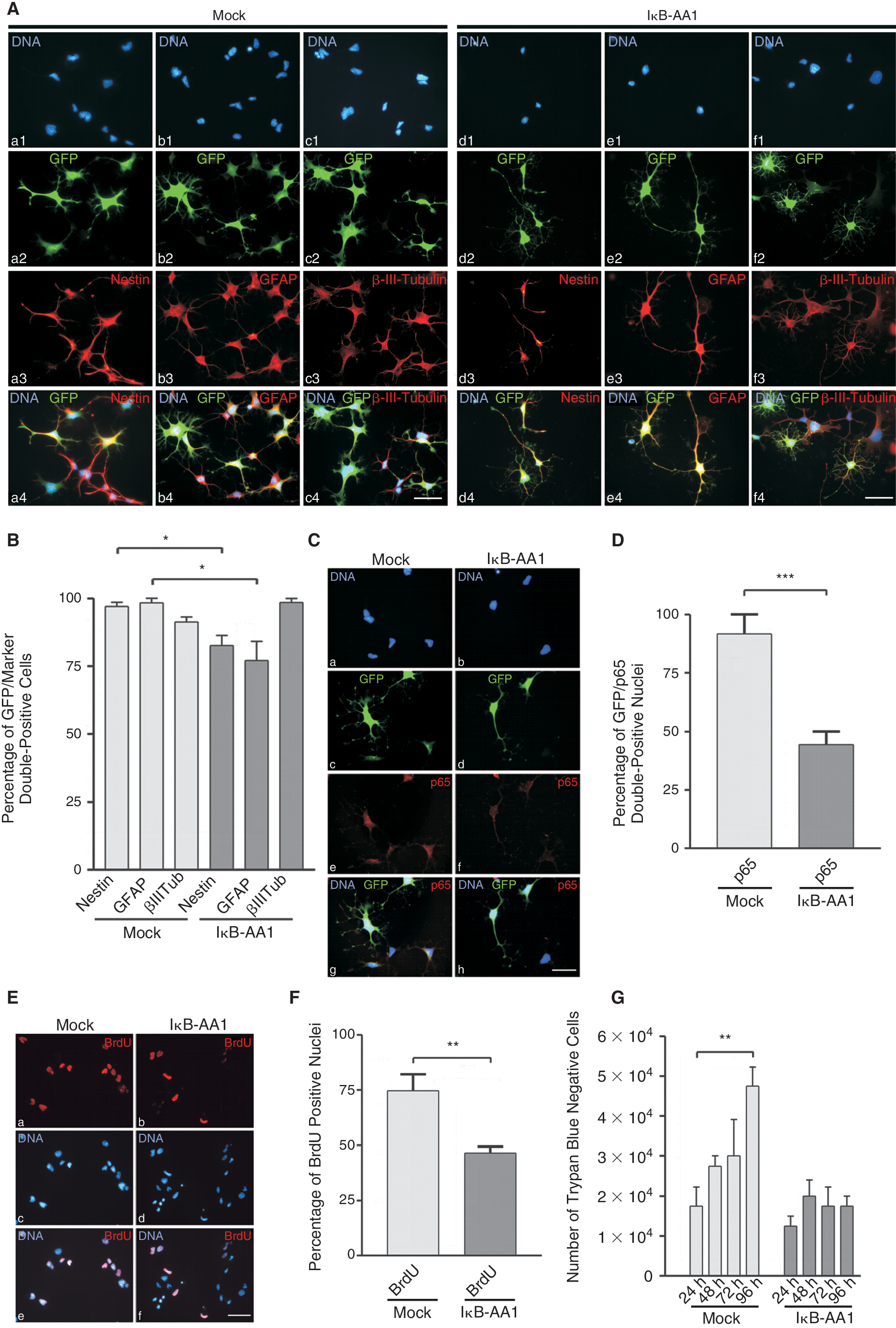
A reduction of constitutively high NF-κB activity in growth factor independent neural stem cells (GiNSCs) reverses the tumorigenic phenotype. (
We quantified proliferation by changes among cell numbers via a trypan blue exclusion assay over a period of 5 days and assessed BrdU incorporation via immunocytochemistry 5 days after differentiation (Fig. 9E). BrdU detection proved nearly 70% of Mock transfected GiNSCs to be highly BrdU positive which was in significant contrast to <50% BrdU positive IκB-AA1 transfected GiNSCs (Fig. 9F). During the neuronal differentiation Mock cell numbers doubled within 96 h whereas IκB-AA1 numbers remained more or less invariable, besides an initial fluctuation (Fig. 9F).
Trypan blue assay was used to determine the amount of living cells (Fig. 9G). When mock transfected GiNSCs proliferated rapidly up to 5 × 10,000 cells within 96 h after seeding of 10,000 cells. In contrast GiNSCs transfected with IκB-AA1 did not proliferate.
Discussion
In our present study we introduce a novel population of NSCs derived from SVZ that are independent of EGF and bFGF supplementation. In the absence of these growth factors these GiNSCs grow as neurospheres. Expression of stemness markers was remarkably similar to control NSCs. They showed to be highly proliferative and had a high constitutive NF-κB activity.
The morphological appearance differed remarkable between GiNSCs and NSCs. In particular, they had enlarged and seemingly decompacted nuclei, pointing at an increased DNA content and a higher transcriptional activity, in which they accumulated the tumor suppressor protein p53. Nuclear p53 without inducing cell death was reported to account for a loss of function and emerging tumorigenesis [43]. Using clinical samples it was shown that a nuclear localization of p53 in 10%–100% of analyzed cells was associated with p53 mutations and tumor progression in bladder cancer. [44] Here we suppose that nuclear p53 in GiNSCs is a sign of nonfunctional mutated p53. In this line a recent study showed that loss of p53 function (p53 –/–) is a major factor for the induction of high-grade malignant gliomas [45]. Additionally, it is very interesting that these gliomas express high levels of VEGF as shown here in GiNSCs [45]. Furthermore conditional expression of mutant p53 in mice resulted in an accumulation of p53 in nuclei of NSCs, similar to GiNSCs, and in the formation of malignant gliomas [46].
GiNSCs even continued to proliferate under differentiation conditions. After inducing neuronal differentiation a markedly persistance of glioma markers Nestin and GFAP was detected. In addition, GiNSCs have an aberrant DNA content, strikingly reduced gene expression levels of neuronal genes and a constitutive NF-κB expression. This type of unregulated NF-κB activity was already reported for many tumors (prostate, breast, lung, lymphomas, see ref. [47]) but is novel for NSC lineages. NF-κB was found to have a driving impact on GiNSC proliferation, as a reduction of its transcriptional activity by the superrepressor IκB-AA1 strongly decreased the proliferation and induced differentiation of the cells. Expression levels of Nestin and GFAP thereby were remarkably reduced and proliferation, measured by independent experimental approaches, decreased as well. The impact of NF-κB on this rescue approach was verified by reduced amounts of activated NF-κB. One of the most stringent state-of-art in vitro assays for detecting malignant transformation of cells is the soft agar assay [48]. It has been demonstrated that the degree of cell growth in semisolid medium consistently correlates with neoplastic growth of cells in nude mice [48]. Here we showed that GiNSCs were able to form colonies in soft agar after growth factor deprivation providing a strong evidence for their potential tumorigenicity. In contrast, nontransformed NSCs formed no colonies.
Cancerous alterations leading to tumors are thought to require initial mutations in proliferating cells. As proliferation is proceeding, NSCs would have the opportunity to accumulate genetic mutations leading to cancerous transformation [49]. In this regard ex vivo expansion of NSCs to obtain sufficient amounts of cells for therapeutic aims, bears the thread of becoming a germ centre for cancerous alterations itself. The here characterized GiNSCs derived from a long-term NSC culture underline this idea, which might add a cautious note for those interested in in vitro propagation of stem cells for therapy. Importantly, long-term culture of stem cells is not only transforming stem cells derived from neural origin as shown here but also leading to spontaneous malignant transformations of bone-marrow derived human mesenchymal stem cells (hMSCs) [50].
The ambivalent role of stem cells in tumorigenesis is underlined by a series of studies supporting the hypothesis of cancerous alteration preferentially occurring in stem cells. A study by Hemmati and colleagues described stem cell properties featuring tumorigenic cells from pediatric brain tumors, which formed neurosphere clusters yielding multipotent and self-renewing cells with the ability to differentiate into neuronal and glial lineages [51].
Taken together optimization of ex vivo culture paradigms is an urgent need for the development of clinical cell transplantation protocols.
Here we provide evidence, that long-term culture has genetically transformed NSCs in a “glioma-like” phenotype. These growth factor-independent GiNSCs have the following cancerous properties: (1) Exogenous growth factor independence; (2) Stemness marker expression as found in human gliomas: Nestin, Musashi, Sox-2; (3) Polyploidity; (4) Nuclear localization of the anti-oncogene p53, without resulting cell death; (5) Transcriptional changes directed by activated NF-κB including upregulation of its target gene VEGF; and (6) Proliferation, dependent on autocrinously produced VEGF.
A recent study linked the genesis of glioma development with aneuploidity and p53 inactivation. They report a clear difference between gliomas and colorectal carcinoma which are diploid or near-diploid [52]. To our knowledge this is the first study reporting on NSC population with genome instability, featuring a constitutively high NF-κB activity, and aberrant differentiation and continuing proliferation.
A line of experimental evidence has assessed the role of NF-κB dependent signaling in different types of cancers. In fact, NF-κB is constitutively active in most tumor specimens described to date and not only blocks apoptosis but also mediates tumor cell proliferation and induces resistance to chemotherapeutic agents [20 –23,53]. In this regard the constitutively high NF-κB activity in GiNSCs and its impact on a glial phenotype and excessive proliferation ignoring differentiation signals might provide a link between NF-κB, cancer and stem cells. Experimental approaches targeting a reduction of NF-κB activity in gliomas have proven promising reduction of proliferation and tumor size [54,55]. The cell of origin of a tumor however and the molecular mechanism, still remains largely unknown. Here we suggest that gliomas might arise from a NSC with genome instability. In accordance with this hypothesis some recent reports provide evidences that NSCs are the cellular origin of high grade gliomas [56,57]. Malignant gliomas are the most aggressive tumors of the brain, which can be clinically treated with drugs like Avastatin or Bevacizumab [58], recombinant humanized monoclonal antibodies against the main proangiogenic factor VEGF [59]. Interestingly, in GiNSC cultures as well, neutralization of VEGF strongly inhibited proliferation and induced differentiation. Interestingly, a constitutive activation of the NF-κB pathway in NSCs using caIKK2 [29] led to increased cell proliferation, growth factor independence and up-regulated VEGF (Fig. 7).
More recently Samuel Weiss and colleagues demonstrated that subpopulations of human glioblastoma stem cells are able to proliferate independently of exogenous mitogens in part through EGFR signaling [60]. Importantly it has been shown that EGF–EGFR interaction is mediated via activation of NF-κB in breast cancer cells [61] (reviewed in ref. [62]).
Thus, we investigated the influence of the NF-κB pathway in U373 human astroglioma cells cultivated as tumorispheres (Supplementary Results). Here we showed that IκB-AA1 transfected glioma cells formed significantly smaller tumorispheres than mock-transfected cells (Supplementary Fig. 1; Supplementary materials are available online at
These data suggest that glioma therapy might be feasible by inhibiting the NF-κB pathway.
Footnotes
Acknowledgments
This study was supported in part by the Deutsche Forschungsgemeinschaft (DFG). The Anti-PSA antibody was obtained from Dr. Christoph Piechaczek, Milteny Biotec, Bergisch Gladbach, Germany. The excellent technical help of Angela Krahlemann-Koehler is gratefully acknowledged.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.