
Abstract
Human embryonic stem (hES) cell differentiation into dopamine neurons is considered a promising strategy for cell replacement therapy in Parkinson's disease, yet the functional properties of hES cell-derived dopamine neurons remain poorly defined. The objective of this study was to characterize intracellular calcium (Ca2+) and sub-plasma membrane cyclic AMP-signaling properties in hES cell-derived dopamine neurons. We found that hES cell-derived dopamine neurons and neural progenitors raised Ca2+ from intra- and extracellular compartments in response to depolarization, glutamate, ATP, and dopamine D2 receptor activation, while undifferentiated hES cells only mobilized Ca2+ from intracellular stores in response to ATP and D2 receptor-induced activation. Interestingly, we also found that hES cell-derived dopamine neurons in addition to primary ventral midbrain dopamine neurons were more prone to release Ca2+ from intracellular stores than non-dopamine neurons following treatment with the neuropeptide neurotensin. Furthermore, hES cell-derived dopamine neurons showed cAMP elevations in response to forskolin and 3-isobutyl-methylxanthine, similar to primary dopamine neurons. Taken together, these results unravel the temporal sequence by which hES cells acquire Ca2+ and cAMP signaling competence during dopamine differentiation.
Introduction
H
Stem cell differentiation protocols are generally evaluated by immunostaining or by polymerase chain reaction (PCR) expression analysis of various key proteins at different time points during the course of development. However, such analysis is insufficient to determine the overall quality of cells, since the functional aspects of proteins and the prevailing function of the cell are not taken into account. Nonetheless, functional methods including neurotransmitter release, electrophysiological recordings of action potentials, and synapse formation have previously been applied to determine the degree of hES cell differentiation [2,3]. Here we describe a new strategy to diagnose the capacity of neurally differentiated hES cells to interact with other cells by responding to neurotransmitter receptor activation with appropriate second messenger signaling.
Parkinson's disease is a neurodegenerative disorder whose principal symptoms, tremor, rigidity, and bradykinesia are mainly caused by the degeneration of mesencephalic dopamine neurons of the substantia nigra. This cell population is responsible for processing several important brain functions, such as motor coordination and planning, attention and learning, as well as reward [4]. To process the tremendous number of functional inputs, and to forward signals to neighboring cells, a network of intricate signaling systems must be in place in the dopamine neurons.
The calcium ion (Ca2+) is a fundamental signaling messenger in all cells [5,6]. As a result of the large concentration gradient over the plasma membrane, Ca2+ can enter the cell via plasma membrane channels and interact with numerous intracellular proteins controlling a vast number of cellular processes. Increase of cytosolic Ca2+ can also take place via release of Ca2+ through intracellular receptors/channels, including the inositol 1,4,5-trisphosphate receptors. Appropriate Ca2+ handling is crucial for the function of all neurons, regulating processes such as exocytosis/endocytosis [7] and synaptic plasticity [8]. cAMP is another fundamental messenger of critical importance for neuronal function [9]. In hES cell-derived dopamine neurons, changes of the intracellular levels of Ca2+ and cAMP are thought to be essential for proper regulation of dopamine release. Nevertheless, Ca2+ and cAMP signaling has not been well described in hES cells at any stage of development toward mature dopamine neurons and remains to be further studied.
Here we demonstrate, by imaging intracellular Ca2+ signaling and sub-membrane cAMP levels, that cell responses change during the course of hES cell differentiation. We found that functional single cell imaging recordings from hES cell derived dopamine cells are similar to primary ventral midbrain dopamine neurons isolated from embryonic mice. In conclusion, our data shed light on the differentiation process of hES cells and provide important functional data that may contribute to: (i) determine the functional state and thus the number of cells to be transplanted in cell replacement therapy and (ii) to develop assays for the screening and development of drugs for Parkinson's disease patients.
Materials and Methods
Human embryonic stem cell cultures
The hES cell lines H9 [1] (46XX, passages 35–55, WiCell license SLA#02-W183) and HS181 [10] (46XX, passages 35–60) were cultured on mitotically inactivated human foreskin fibroblasts (hFF; ATCC; CRL-2429) in a medium containing: KO-DMEM medium (Invitrogen, Carlsbad, CA), 20% knockout serum replacement (SRM; Invitrogen), 2 mM
Neural induction and differentiation
Neural differentiation of hES cells was induced by means of co-culture on PA6 stromal cells as described previously [2,11]. In brief, hES cells (2 × 104 cells per 6 cm culture dish) were plated on a confluent layer of PA6 cells (mitotically inactivated) in SRM containing KO-DMEM, 15% knockout serum replacement, 2 mM
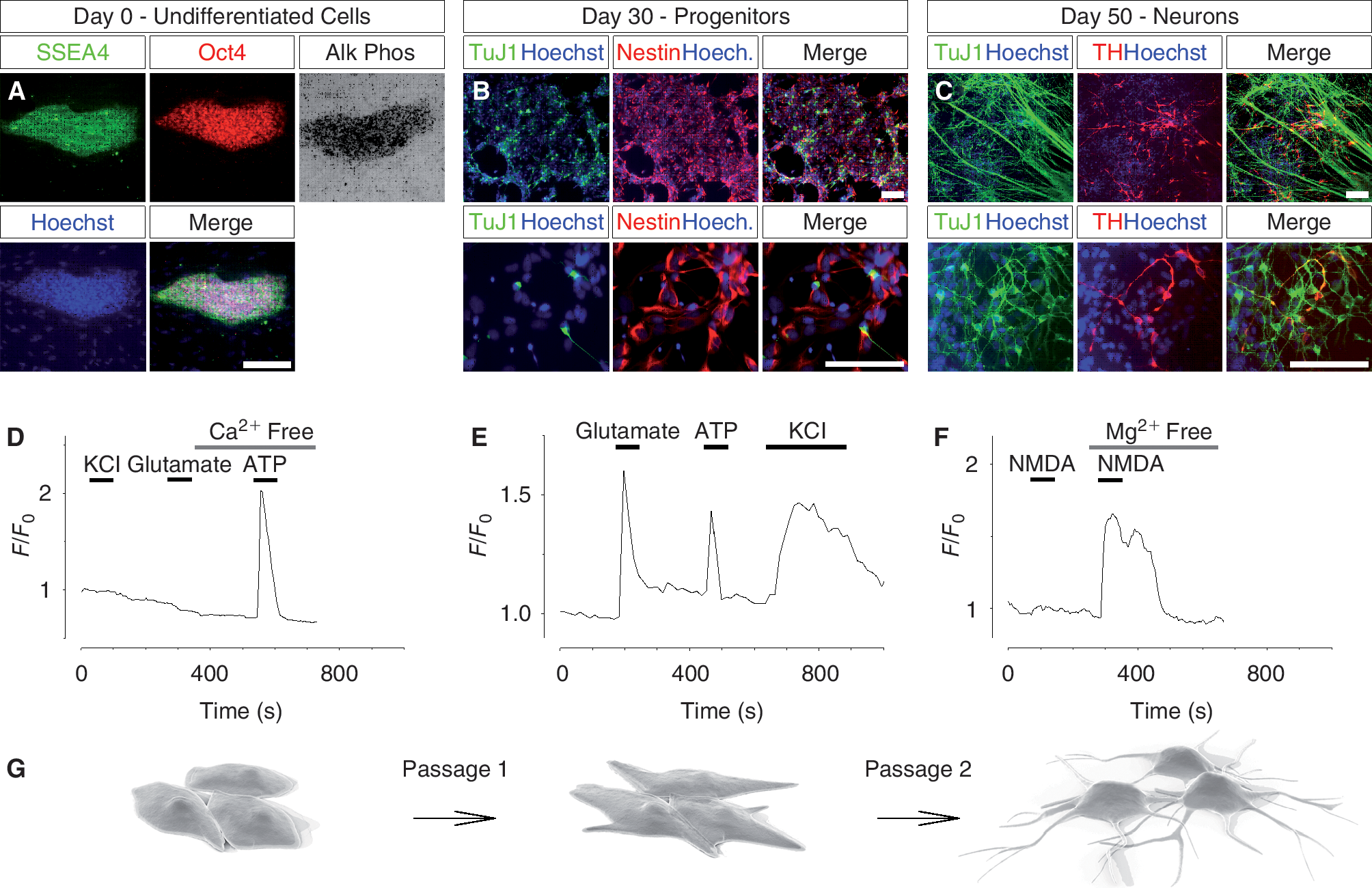
Differentiation of human embryonic stem (hES) cells into dopamine neurons and Ca2+ responses at various stages of differentiation. (
Primary culture of dopamine neurons
Embryonic mouse (E12.5) ventral mesencephala of CD1 (wild type) or c57BL/6 expressing GFP under the control of the TH promoter [12,13] (TH-GFP) were manually dissected, dissociated, and plated as previously described [14]. In brief, primary cells (1 ×105 per well) were cultured on polyornithine/laminin 24-well plates and then grown in serum-free N2 medium supplemented with Shh, FGF8, BDNF, and AA (using similar concentrations as above) for 3–4 days.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde for 20 min, blocking with 5% serum and 0.25% triton and incubated overnight with the following antibodies: mouse monoclonal antibodies: β-tubulin III (TuJ1; 1:1,000; Promega, Madison, WI); TH (1:300; Diasorin, Saluggia, Italy); SSEA4 (1:100; Developmental Studies Hybridoma Bank, University of Iowa, IA); and/or rabbit polyclonal antibodies: TH (1:500; Pel-Freez Biologicals, Rogers, AR); Nestin (1:1,000; Chemicon, Temecula, CA); Oct4 (1:300; Santa Cruz Biotechnology, Santa Cruz, CA). Donkey anti rabbit Cy3- and goat anti-mouse Cy2-labeled (Jackson ImmunoResearch Laboratories, West Grove, PA) secondary antibodies were used. Cell nuclei were visualized by Hoechst 33258 (Invitrogen) counterstaining. Alkaline phosphatase activity, in undifferentiated hES cells, was detected with Alkaline Phosphatase Kit (Vector Laboratories, Burlingame, CA) as recommended by the manufacturer.
Intracellular Ca2+ imaging
Cells were loaded with the Ca2+-sensitive fluorescence indicator Fluo-3/AM or Fura-2/AM (Invitrogen) at a concentration of 5 μM in cell culture medium at 37°C for 30 min. The Ca2+ measurements were carried out at 37°C in a heat-controlled perfusion chamber (QE-1; Warner Instruments, Hamden, CT) with a cooled back-illuminated EMCCD camera (Cascade II; Photometrics, Tucson, AZ) mounted on an inverted microscope (Zeiss Axiovert 100M; Carl Zeiss, Jena, Germany) equipped with a 25×/0.8NA water objective (Carl Zeiss). Excitation at 495 nm was carried out using a Lambda LS xenon-arc lamp equipped with a SmartShutter (Sutter Instrument, Novato, CA). Emission wavelengths were detected at 510 nm and the sampling frequency was set to 0.2–1 Hz. Experiments were performed in a Krebs-Ringer's buffer containing 119.0 mM NaCl, 2.5 mM KCl, 2.5 mM CaCl2, 1.3 mM MgCl2, 1.0 mM NaH2PO4, 20.0 mM HEPES (pH 7.4), and 11.0 mM dextrose. In experiments with zero extracellular Ca2+ concentration, CaCl2 was substituted with 2.5 mM EGTA. The computer software MetaFluor (Molecular Devices, Sunnyvale, CA) was used to control all devices and to analyze acquired images. Cells were grown on gridded coverslips to enable identification of specific cell types after single cell Ca2+ recordings. Gridded coverslips are photo-etched on the surface for a permanent pattern containing 200 numbered squares, each 500 μm width (Electron Microscopy Sciences, Hatfield, PA). When performing time-lapse microscopy experiments, a bright-field image of the etched coordinates at the field of view was recorded. After immunostaining with specific neural markers (see Immunocytochemistry), the same field of view was localized and protein expressions were correlated with intracellular Ca2+ responses.
Measurement of intracellular cAMP in single cells
The cAMP concentration in the sub-plasma membrane space was measured with total internal reflection fluorescence (TIRF) microscopy and a fluorescent biosensor as previously described [15]. In brief, the full-length PKA catalytic Cα subunit was labeled with yellow fluorescent protein (Cα-YFP) and a truncated PKA-RIIβ regulatory subunit was labeled with cyan fluorescent protein and targeted to the plasma membrane via a covalent lipid modification (ΔRIIβ-CFP-CAAX). The two constructs were nucleofected into primary dopamine neurons and hES cell-derived dopamine neurons, which were then plated on glass coverslips. Nucleofection was performed according to the supplier's recommendations (Amaxa, Lonza, Basel, Switzerland). Plasma membrane association of the fluorescent protein-tagged PKA subunits were measured using a custom-built TIRF microscope setup [16] at 37°C in a heat-controlled perfusion chamber mounted on the stage of an upright microscope (E600FN; Nikon, Tokyo, Japan) equipped with a 40×/0.8NA water immersion objective (Nikon). Excitation at 442 and 514 nm were carried out using a helium–cadmium (Kimmon, Tokyo, Japan) and an argon laser (Creative Laser Production, Munich, Germany) with the beams focused through a modified quartz dove prism (Axicon, Minsk, Belarus) at an angle of 70° to achieve total internal reflection. Emission wavelengths were selected with interference filters (485 nm/25 nm half-bandwidth for CFP and 560/40 nm for YFP; Semrock, Rochester, NY) mounted in a filter wheel (Sutter Instruments) and fluorescence recorded with a back-illuminated EMCCD camera (DU-887; Andor Technology, Belfast, Northern Ireland). The computer software MetaFluor (Molecular Devices) was used to control all devices and to analyze acquired images. Sampling frequency was set to 0.2 Hz. Experiments were performed in a Krebs-Ringer's buffer. cAMP concentration was expressed as the ratio of CFP over YFP fluorescence after background subtraction.
Reagents
Drugs were purchased from Sigma, except for N-methyl-
Data analysis
The Ca2+ recording data was normalized and cells were considered responsive to a treatment if the mean fluorescent was increased by at least 25% over the baseline. The data are presented as the means ± SEM. The Student's t-test was used and significance was accepted at P < 0.05.
Results
Human embryonic stem cells develop an intact Ca2+ signaling machinery during neural differentiation
To explore the functional properties of embryonic stem cells during the process of neural differentiation, we differentiated hES cells toward dopamine neurons as previously described [2]. Cells were analyzed at 3 different stages (Fig. 1G): in the undifferentiated embryonic stem cell stage (day 0; Fig. 1A), during neural differentiation (day 30; Fig. 1B), and at the end of the differentiation period (day 50; Fig. 1C).
The cell functionality at various stages of neural differentiation was determined by performing single cell live imaging of the intracellular Ca2+ concentration. Undifferentiated hES cells, found to express SSEA4 and Oct4 plus alkaline phosphatase activity (Fig. 1A), were loaded with the Ca2+-sensitive dye Fluo-3/AM and challenged with different stimuli known to induce intracellular Ca2+ signaling. Neither depolarization with 50 mM KCl nor 100 μM glutamate triggered a Ca2+ response in undifferentiated hES cells (Fig. 1D). When cells were exposed to 10 μM ATP, however, a robust Ca2+ transient was observed (Fig. 1D). The Ca2+ response persisted in the absence of extracellular Ca2+, indicating an involvement of internal Ca2+ stores. Correspondingly, hES cells differentiated for ∼14 days responded to ATP, but not to glutamate or KCl (data not shown). After 28 days of differentiation on stromal cells, rosette structures containing neural progenitor cells were manually isolated and replated without feeder cells. Two days later (day 30), the neural progenitor cells expressed nestin (Fig. 1B). When these cells were loaded with the Ca2+ indicator and imaged, Ca2+ responses were detected not only after treatment with 10 μM ATP, but also in response to 100 μM glutamate and 50 mM KCl (Fig. 1E). Percentage responding cells were 80.3% ± 8.7% (glutamate), 75.6% ± 15.1% (ATP), and 89.3% ± 8.6% (KCl; n > 150, N > 3). Fully differentiated hES cell-derived dopamine neurons (day 50) were examined with typical neural/dopamine markers and found positive to: β-tubulin III (TuJ1), tyrosine hydroxylase (TH), VMAT, Girk2, DAT, Engrailled, Lmx1a, Nurr1, and negative to: Nestin, 5-HT, GABA (Supplementary Fig. 1; supplementary materials are available online at
Next we examined intracellular Ca2+ responses when treating the fully differentiated neurons (day 50) with the well-known neurotransmitters AMPA and DHPG. To back-trace cells that were subjected to Ca2+ recordings, we used gridded coverslips (Fig. 2A). Applying this technique, we could immunostain neurons derived from hES cells for various neuronal markers after the Ca2+ recordings and correlate the expression with detected Ca2+ responses. Using this approach, we immunostained the cells for TuJ1 and TH (Fig. 2B). Fully differentiated TuJ1+/TH+ neurons (day 50) responded to 10 μM AMPA (Fig. 2C and 2D) or 1 μM DHPG (Fig. 2E and 2F) with a Ca2+ transient. DHPG activates metabotropic glutamate receptors that trigger the release of Ca2+ from intracellular Ca2+ stores, while AMPA activates the ionotropic AMPA receptors that elevate the cytosolic Ca2+ concentration through influx of Ca2+ from the extracellular milieu. As expected, DHPG still evoked a Ca2+ response in the absence of extracellular Ca2+, whereas AMPA or KCl did not (Fig. 2E). There was no significant difference in percentage responding cells, Ca2+ transient amplitude, or full duration at half maximum (FDHM), when comparing AMPA and DHPG responses in non-dopamine neurons (TuJ1+/TH−) with dopamine neurons (TuJ1+/TH+; Fig. 2D and 2F).
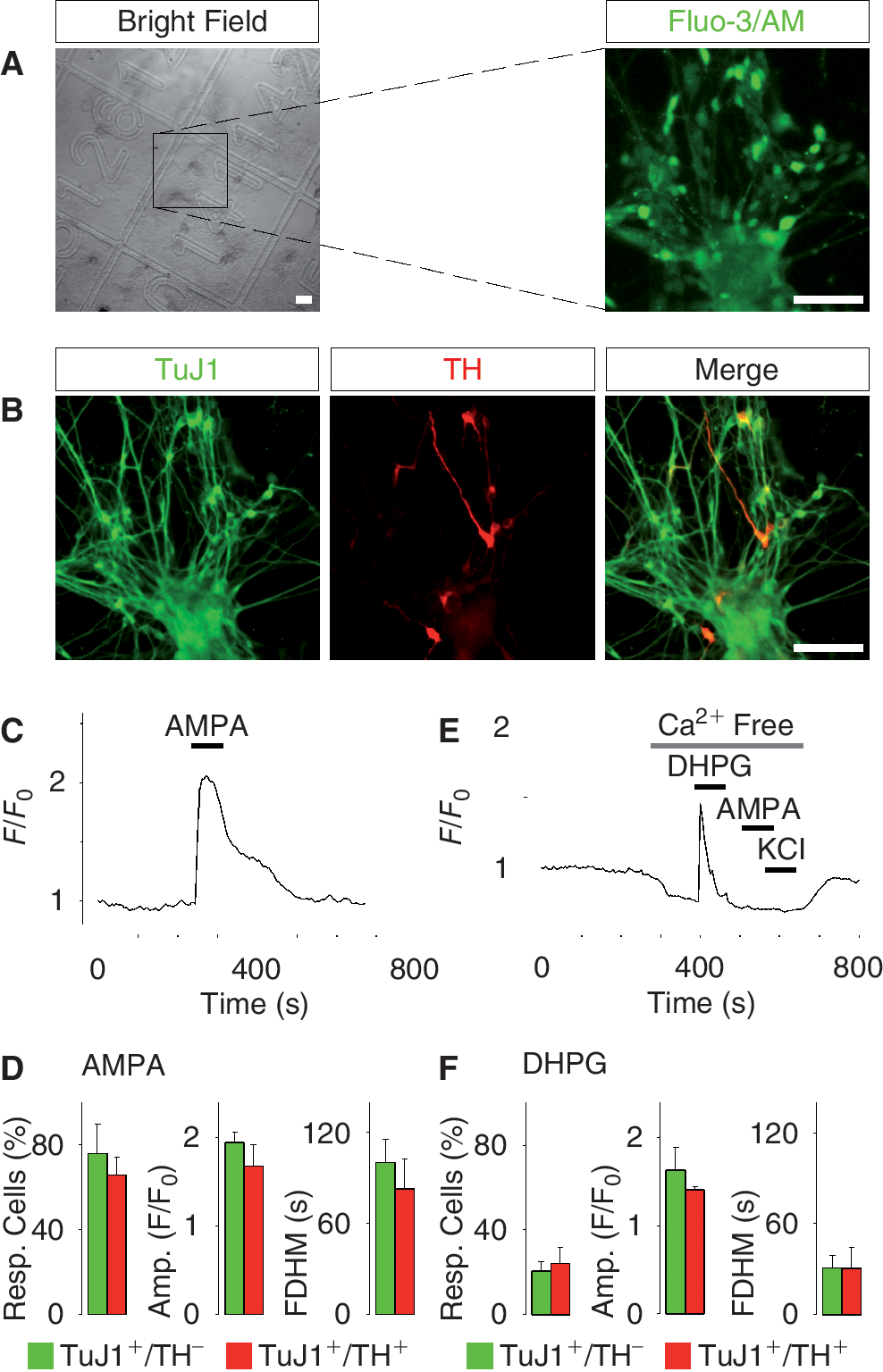
Ca2+ responses to various agonists in fully differentiated human embryonic stem (hES) cell-derived dopamine neurons. (
Neurotensin triggers Ca2+ signaling in human embryonic stem cell-derived dopamine neurons
The similar Ca2+ responses to general neurotransmitters in non-dopamine neurons (TuJ1+/TH−) and dopamine neurons (TuJ1+/TH−) prompted us to examine whether hES cell-derived dopamine neurons are also capable of responding to specific neurotransmitters of dopamine neurons. To address this issue, we examined the capacity of hES cell-derived dopamine neurons to respond to NT with Ca2+ signaling. NT is a neuropeptide that activates the NT receptor and triggers Ca2+ release from intracellular stores, which results in increased firing rate of dopamine neurons [17]. Fully differentiated TuJ1+/TH− neurons (day 50) derived from hES cells (Fig. 3A) responded with a prominent Ca2+ transient when exposed to 1 μM NT in the absence of extracellular Ca2+ (Fig. 3B and 3C), which indicate that the neurotransmitter released Ca2+ from internal stores. The percentage of TuJ1+/TH+ neurons responding to NT was significantly higher, as compared with TuJ1+/TH− cells (59.4% ± 6.0% and 25.6% ± 7.0%, respectively, P < 0.05; Fig. 3D), but there was no significant difference between the amplitudes and FDHM of NT-induced transients (TuJ1+/TH+ cells n = 34, TuJ1+/TH− cells n = 88, N = 3; Fig. 3D). The Ca2+ transient induced by NT was specific, since inhibiting the NT receptor with 200 nM SR142948 effectively blocked the Ca2+ response evoked by NT, but not the response triggered by DHPG (Fig. 3E). Control experiments showed that NT and DHPG sequentially induced a Ca2+ response in the TuJ1+/TH+ cells (Fig. 3F). In sum, these data show that TuJ1+/TH+ neurons derived from hES cells differ from TuJ1+/TH− cells in that they are more prone to respond to NT.
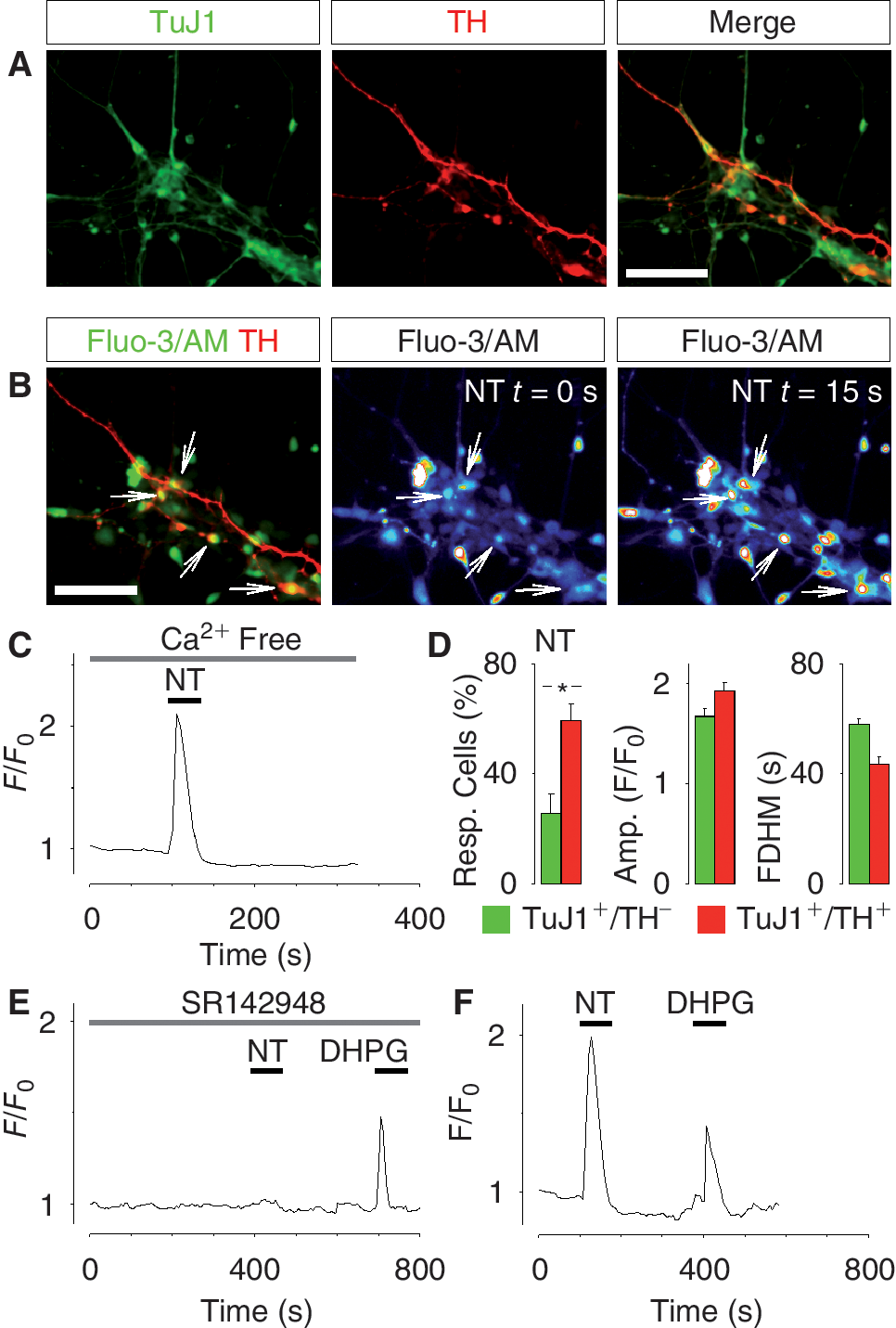
Neurotensin (NT)-induced Ca2+ signaling in fully differentiated human embryonic stem (hES) cell-derived dopamine neurons. (
Human embryonic stem cell-derived dopamine neurons show similar functional responses as primary ventral midbrain dopamine neurons
We next set out to compare in vitro differentiated hES cell-derived dopamine neurons with in vivo differentiated dopamine neurons isolated from mouse ventral midbrain. In order to study the regulation of Ca2+ signaling in ventral midbrain dopamine neurons, we prepared primary cultures using wild-type mice and a genetically modified mouse strain expressing GFP under the TH promoter. After 3 days in culture, ventral midbrain dopamine neurons were identified by TH immunostaining (Fig. 4A). Neurons derived from the TH-GFP transgenic mice additionally showed a strong GFP signal (Fig. 4B). Primary dopamine neurons were loaded with the Ca2+ indicator Fluo-3/AM or Fura-2/AM (Fig. 4A and 4B) and challenged with various agonists provoking a rise in the intracellular Ca2+ concentration. The response to 10 μM AMPA in TH+ cells and TH− cells was similar in all parameters examined (Fig. 4C) and did not differ from that in hES cell-derived dopamine neurons (Fig. 3). Interestingly, we found that ventral midbrain TH+ neurons were more prone to respond to 1 μM NT compared with TH− neurons (30.1% ± 2.2% vs. 9.3% ± 2.8%, P < 0.05), but among responding cells the amplitude or FDHM of the Ca2+ transient did not differ (Fig. 4D). Moreover, like hES cell-derived neurons, primary dopamine neurons responded with a prominent Ca2+ transient when exposed to 1 μM NT in the absence of extracellular Ca2+ (Fig. 4E). Inhibiting the NT receptor with 200 nM SR142948 effectively prevented the NT-evoked Ca2+ response in primary dopamine neurons, but did not affect the Ca2+ release from internal stores triggered by 1 μM DHPG (Fig. 4F). Control experiments showed that NT and DHPG sequentially induced a Ca2+ response in primary dopamine neurons (Fig. 4G). These results indicate that the functional properties of dopamine neurons obtained by differentiation of hES cells are similar to the properties of primary dopamine neurons.
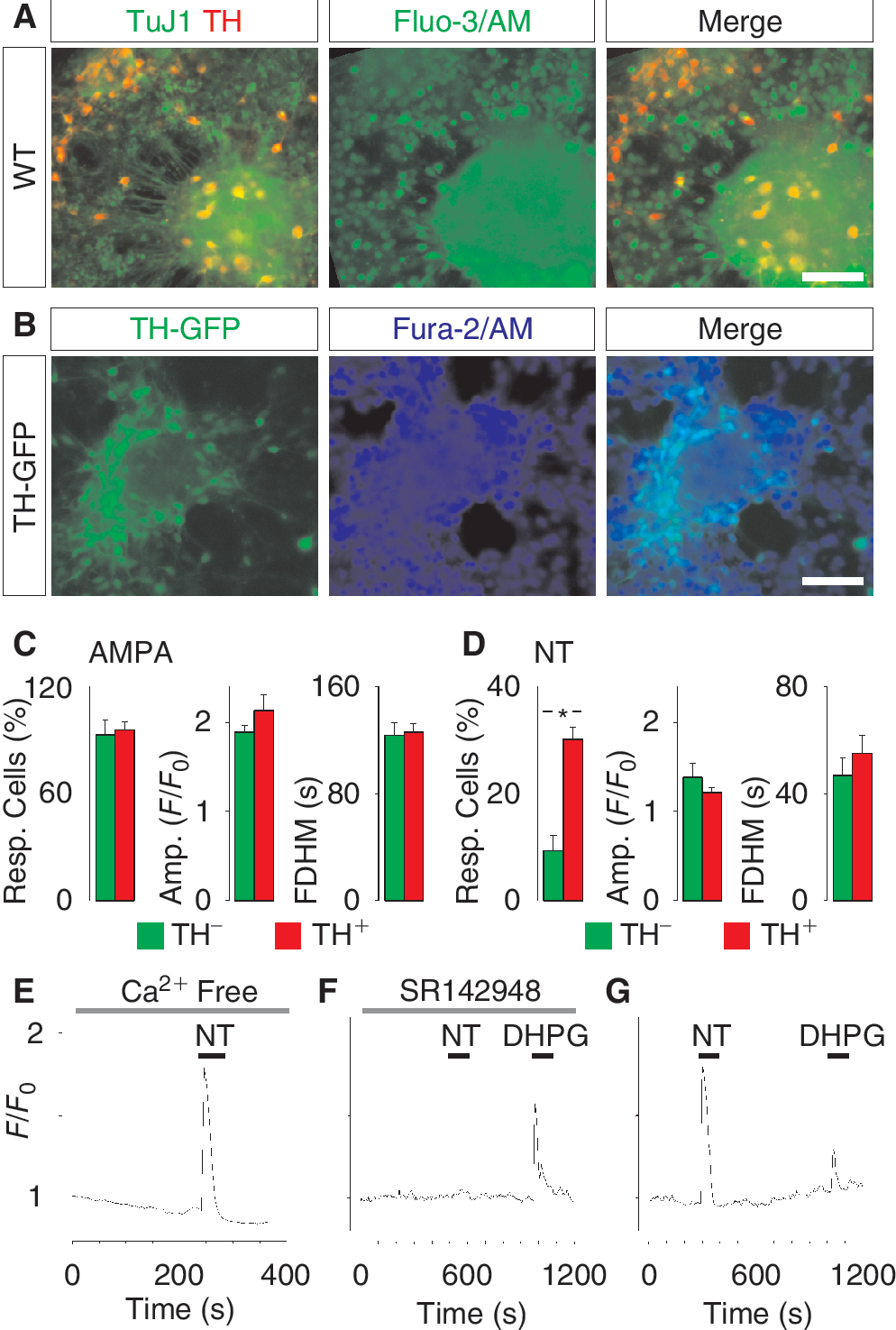
Ca2+ signaling in primary ventral midbrain dopamine neurons isolated from embryonic mice. (
Dopamine D2 receptor activation triggers intracellular Ca2+ signaling in dopamine neurons
The dopamine signaling system is implicated in the regulation of multiple cellular functions in different types of cells [18,19]. To explore if specific activation of dopamine receptors could trigger a response, we examined hES cell-derived dopamine neurons by Ca2+ imaging, using primary dopamine neurons as well as undifferentiated hES cells as control. Treatment of Fluo-3/AM loaded hES cell-derived dopamine neurons with 100 μM quinpirole, a specific agonist of the D2 subclass of receptors, evoked a strong Ca2+ transient (Fig. 5A). This Ca2+ response persisted even in the absence of extracellular Ca2+ (Supplementary Fig. 3A), suggesting that intracellular Ca2+ stores played a role in this signaling event. The dose dependency of the quinpirole-induced Ca2+ response was tested by exposing the cells to 1, 10, and 100 μM quinpirole (Supplementary Fig. 3B). Analyzing the percentage responding cells, amplitude and FDHM showed no significant difference between TuJ1+/TH+ cells and TuJ1+/TH− cells treated with 100 μM quinpirole (TuJ1+/TH+ cells n = 64, TuJ1+/TH− cells n = 137, N = 3; Fig. 5B). Unexpectedly, 100 μM quinpirole also evoked a Ca2+ response in the majority (>73%, n = 200) of the undifferentiated hES cells (day 0; Fig. 5C). The response to specific D2 receptor activation was next examined in in vivo differentiated dopamine neurons isolated from mouse ventral midbrain. Treating primary dopamine neurons with 100 μM quinpirole triggered Ca2+ transients (Fig. 5D). These data indicate that the ability to signal through the D2 receptor with Ca2+ signaling is a fundamental property in both undifferentiated hES cells and differentiated dopamine neurons generated in vitro or in vivo.
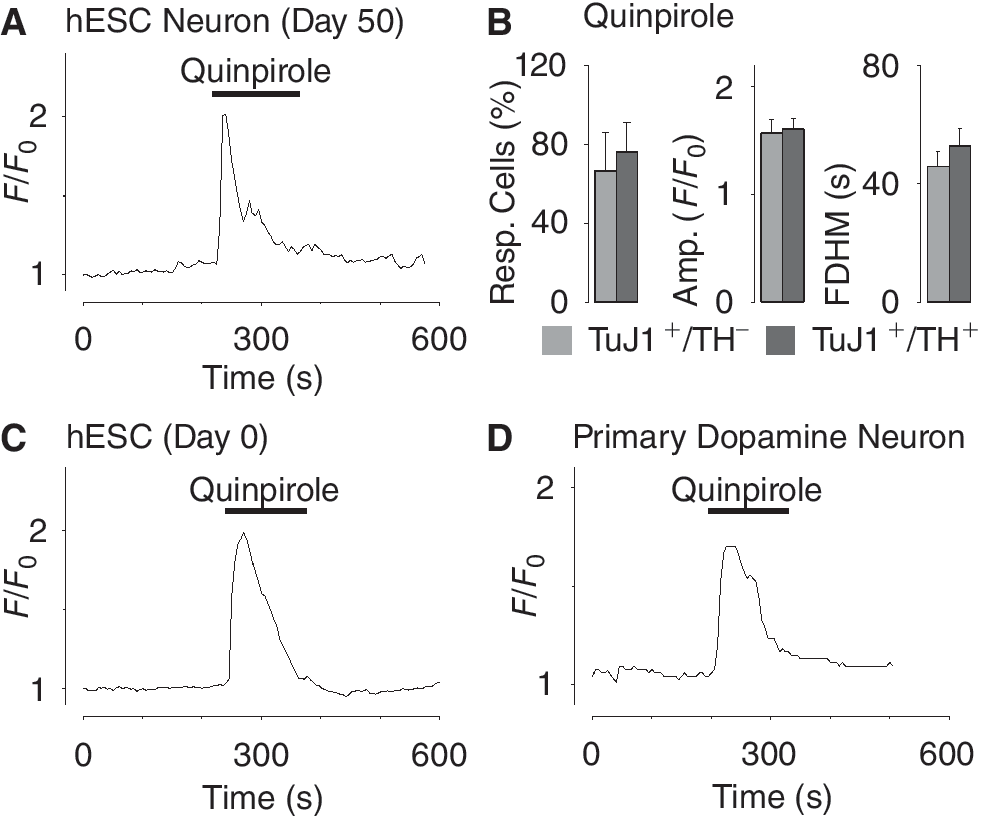
Dopamine-induced Ca2+ signaling in fully differentiated human embryonic stem (hES) cell-derived dopamine neurons, undifferentiated hES cells, and primary dopamine neurons. (
Human embryonic stem cell-derived dopamine neurons have intact cAMP signaling
As dopamine neurons possess dopamine autoreceptors that signal via cAMP, we examined the functionality of this pathway [20,21] in hES cell-derived dopamine neurons. The intracellular cAMP signaling machinery was examined using a fluorescent translocation probe for cAMP. hES cell-derived dopamine neurons differentiated for 50 days were nucleofected with plasmids encoding a CFP-tagged PKA regulatory subunit, that was truncated and membrane targeted, and a YFP-tagged PKA catalytic subunit [15] (Fig. 6A and 6B). Cells were imaged with a custom-built TIRF microscope [16] during exposure to the adenylate cyclase activator forskolin and the phosphodiesterase inhibitor isobutyl-1-methylxanthine (IBMX). Cells treated with 10 μM forskolin showed a marked decrease in plasma membrane YFP signal (Fig. 6C). The resulting increase of the CFP/YFP ratio corresponds to an increase in the sub-plasma membrane concentration of cAMP (Fig. 6C). This response was observed in 30.9% 5.5% of the cells (n > 300, N = 4). Similarly, 100 μM IBMX also triggered a decrease in the YFP signal (Fig. 6D) and increased the cAMP-dependent CFP/YFP ratio. These results show that hES cell-derived dopamine neurons are not only capable to respond with Ca2+ signaling, but also possess the basic components of the cAMP signaling machinery.
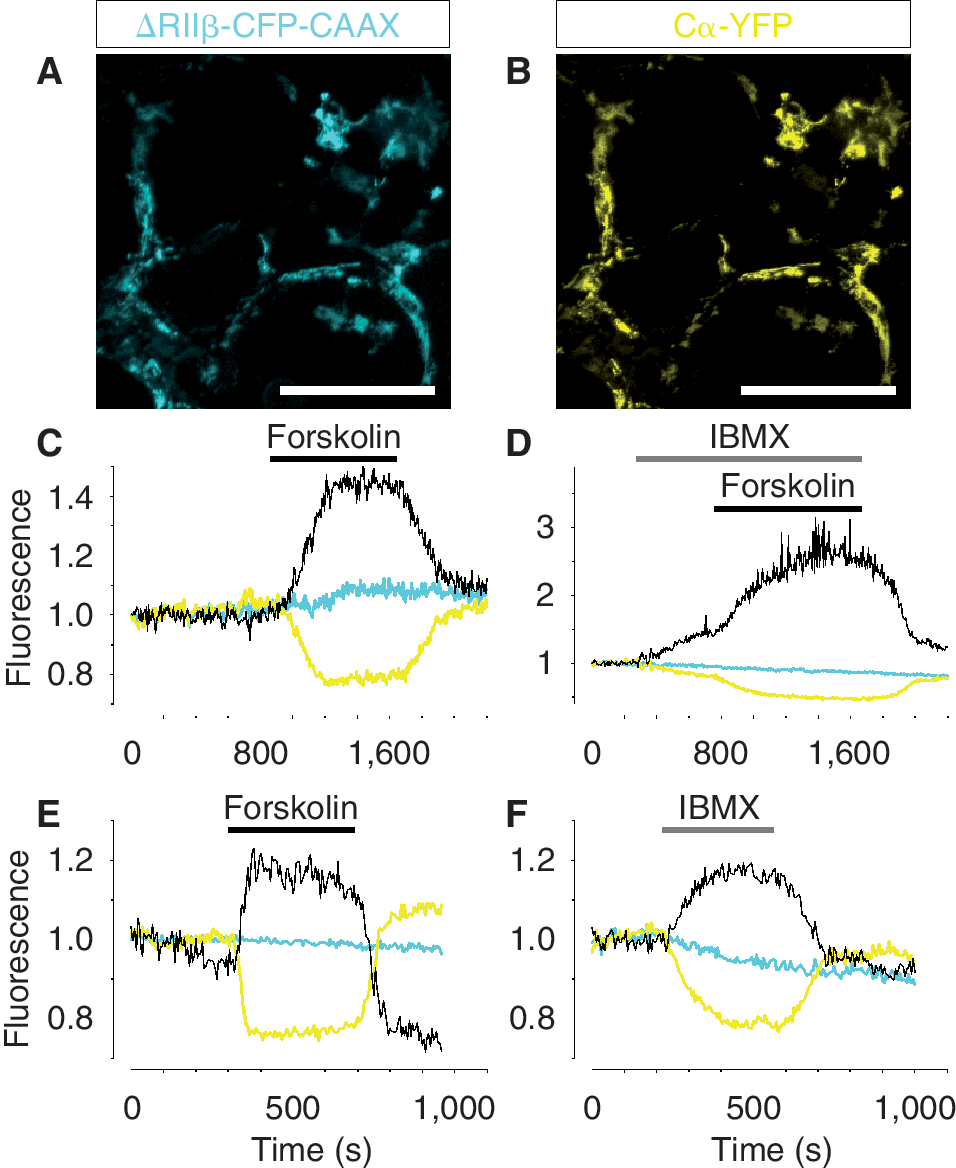
cAMP signaling in fully differentiated human embryonic stem (hES) cell-derived neurons and primary dopamine neurons. (
Finally, we examined whether primary dopamine neurons isolated from wild-type mice responded to drugs that modulate cAMP signaling in a similar way as hES cell-derived dopamine neurons. Primary dopamine neurons were nucleofected with the translocation biosensor for sub-membrane cAMP recordings. Indeed, we observed that 10 μM forskolin (Fig. 6E) and 100 μM IBMX (Fig. 6F) induced cAMP elevations similar to those observed in hES cells. Thus, in sum, our data show that dopamine and non-dopamine neurons derived from hES cells have cAMP and Ca2+ signaling properties resembling those of primary dopamine and nondopamine neurons isolated from embryonic mice.
Discussion
In this study, we explored the functional properties of hES cell-derived dopamine neurons. To this end, measurements of the intracellular Ca2+ concentration and levels of cAMP were conducted in living cells. Our data reveal that undifferentiated hES cells lack fundamental properties of the Ca2+ signaling machinery and that differentiating hES cells progressively acquired Ca2+ signaling competence. Neural progenitor cells and hES cell-derived dopamine neurons show Ca2+ and cAMP signaling patterns similar to primary ventral midbrain dopamine neurons isolated from embryonic mice.
Ca2+ and cAMP signaling in dopamine neurons derived from human embryonic stem cells
Ca2+ and cAMP are two ubiquitous messengers that regulate numerous cellular processes [9,22]. In neurons, cAMP plays a critical role in facilitating synaptic transmission and regulating synaptic plasticity associated with learning and memory [9]. Signaling via Ca2+ is one of the most general signaling mechanisms in cells. It exists in all cell types, at all developmental stages and regulates a wide variety of cell functions that range from fertilization to cell death [23]. Our study demonstrates that NT receptor stimulation triggers intracellular Ca2+ release in dopamine neurons obtained from hES cells in vitro and from the ventral midbrain in vivo. In addition, we demonstrate D2 receptor-activated Ca2+ responses and forskolin-triggered cAMP signaling in hES cell-derived dopamine neurons as well as in primary dopamine neurons. Thus, our data show that dopamine neurons derived from hES cells are capable of responding to cell type-specific stimuli activating both the cAMP and Ca2+ signaling machinery in an appropriate manner and to a similar extent as in primary dopamine neurons.
Ca2+ signaling in differentiating human embryonic stem cells
Undifferentiated hES cells, unlike neuralized hES cells, did not respond to depolarization nor to stimulation with excitatory amino acids, indicating that voltage-gated Ca2+ channels and certain neurotransmitter receptors do not become functional until later stages of differentiation. However, undifferentiated hES cell responded to ATP and D2 receptor activation with releasing Ca2+ from intracellular stores, suggesting that these signaling mechanisms are essential at very early stages of development. Indeed, Ca2+ signaling already plays an important role at the fertilization stage when a Ca2+ wave is triggered by the sperm fusing to the egg, forming the zygote [24,25]. Moreover, our experiments show that hES cells respond with intracellular Ca2+ release throughout the entire process of differentiation into neurons, suggesting that intracellular Ca2+ release is a fundamental and conserved mechanism during development. Our data also reveal that the ability of hES cells to respond to various Ca2+ signaling stimuli increases significantly upon neural differentiation. Importantly, neural progenitor cells derived from isolated neural rosettes responded with Ca2+ signaling both to KCl depolarization and glutamate stimulation, indicating that both voltage-gated channels and neurotransmitter receptors are functional at this stage of differentiation. Furthermore, a similar type of response was observed when hES cell-derived dopamine neurons were examined after 50 days of differentiation. Thus, as cells differentiate into neural progenitors and dopamine neurons, they acquire a more sophisticated signaling machinery that allow them to mobilize Ca2+ from the extracellular space and respond to multiple neurotransmitters. An example of the latter would be the regulation of Ca2+ signaling by NT, which distinguished dopamine neurons from other neurons.
Implications for development of novel treatment modalities for Parkinson's disease
Human pluripotent stem cells are considered as the ultimate source of cells in regenerative medicine [26,27]. Indeed, hES cells have been proposed as therapeutic tools for cell replacement therapy in neurodegenerative disorders such as Parkinson's disease. Research has thus focused on the derivation of dopamine neurons from hES cells [2,28,29] with the goal of transplanting patients suffering from Parkinson's disease. At the same time progress in the generation of hES cell-derived dopamine neurons and in the generation of induced pluripotent stem (iPS) cells [30] has provided a momentous impetus both to the development of cell replacement therapies [31] and to the development of human cell-based assays for drug discovery, toxicology, and drug development [26 –34]. Moreover, since iPS cells can also be derived directly from patients, it is expected that such cells may become ideal tools to model disease, as was recently reported for familial dysautonomia [35]. These achievements have generated a lot of interest in the pharmaceutical industry as they opened the door for the development of assays for drug screening in normal and diseased human cells at an unprecedented scale. There is therefore a growing need to functionally characterize the numerous new cell lines currently being generated, and at the same time a demand for understanding the signaling mechanisms in dopamine neurons to validate such mechanisms in hES cell-derived dopamine neurons. Here we demonstrate that Ca2+ imaging is an instantaneous and efficient method to characterize cell types and study their signaling mechanisms. Ca2+ signaling has so far, to our knowledge, only been characterized in mouse embryonic stem cell-derived neurons [36,37] and very little is known about the functional characteristics of hES cell-derived dopamine neurons or how they compare with primary dopamine neurons. In this context, our results provide evidence that the functional properties of hES cell-derived dopamine neurons are, in all we have examined, similar to primary dopamine neurons. These results validate the method and technology used to analyze the functional properties of hES cell-derived dopamine neurons. We therefore think that the use of this approach can be further extended to study other aspects of hES cell- or pluripotent cell-derived progeny, including dopamine neurons. In the case of hES cell-derived dopamine neurons, the approach used in this study can be adapted to high-throughput screenings (HTS) aiming at identifying drugs that modulate Ca2+- or cAMP-dependent functions. Finally, our results suggest that techniques such as those applied here could be used to characterize and adjust the functional status and capacity of cell preparations for cell replacement therapies.
Conclusion
By imaging intracellular Ca2+ signaling and sub-membrane cAMP levels, we have found that cell signaling responses change during the course of hES cell differentiation. Furthermore, we demonstrate that TH+ cells differentiated from hES cells have similar functional properties when compared with primary ventral midbrain dopamine neurons isolated from embryonic mice. Our data shed light not only on the differentiation process of stem cells, and on the functional properties of hES cell-derived dopamine neurons, but also on the approach to functionally evaluate single cells and develop single cell assays relevant for future development of HTS systems and stem cell therapies in regenerative medicine.
Footnotes
Acknowledgments
This study was supported by Vetenskapsrådet—the Swedish Research Council (DBRM), Knut and Alice Wallenberg Foundation (CLICK and Research Fellow), The Royal Swedish Academy of Sciences, the Foundation for Strategic Research (CEDB), Åke Wiberg's Foundation, Jeansson's Foundation, Magnus Bergval's Foundation, Fredrik och Ingrid Thuring's Foundation, the Swedish Society for Medical Research, Family Ernfors' Foundation, Novo Nordisk Foundation, and the Swedish Diabetes Association. The authors wish to thank Dr. Ruani Fernando and the staff of the Molecular Neurobiology Unit, Department of Medical Biochemistry and Biophysics.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.