
Abstract
Adult human dental pulp contains stem cells (DPSCs) that are capable of differentiation into osteoblasts, odontoblasts, adipocytes, and neuronal-like cells. Because these cells have potential use in tissue regeneration, herein we characterized the response of DPSC lines to ionizing radiation (IR). These DPSC lines have been developed from the extracted molars of healthy donors. DPSCs were cultivated in a unique media supplemented with epidermal growth factor (EGF) and platelet-derived growth factor (PDGF). Since tissue homeostasis depends on a precise balance among cell proliferation, senescence, and cell death, we explored the effects of IR (2–20 Gy) on the proliferative activity of DPSCs and the molecular pathways involved. Even the highest dose used (20 Gy) did not induce DPSC apoptosis. After irradiation with doses of 6 and 20 Gy, DPSCs accumulated in the G2 phase of the cell cycle. DPSCs responded to IR (20 Gy) with senescence detected as SA-β-galactosidase positivity, beginning on the third day after irradiation. Twenty-four hours after irradiation, p53 and its serine 15 and 392 phosphorylated forms were detected. At this time, p21 (WAF1) was induced. Increases in protein p16 were observed from the third day following irradiation and continued till the end of the examination (Day 13). We conclude that DPSCs respond to IR-induced damage by permanent cell cycle arrest in the G2 phase and by stress-induced premature senescence.
Introduction
S
SCs are equipped with other mechanisms contributing to SC resistance, such as a generally high resistance to stress with an increased DNA repair capacity, a high amount of molecular chaperons and well-developed ubiquitin-proteasome and detoxification systems [9].
We may question how SCs react to genotoxic stressors, such as ionizing γ irradiation, used in the treatment of many cancer types. IR causes severe damage to DNA, including single- and double-strand breaks (DSB). So far, the situation is quite well mapped for HSCs. The cells react to DNA damage caused by IR by cell cycle arrest and transient arrest in the G1/S or G2/M phase, where they repair the damage. The repair takes time, and thus highly active cells are much more susceptible to IR than quiescent cells. In the case of human hematopoietic CD133+ cells irradiated in vitro, the cells have decreased clonogenic capacity, and if the damage is not repaired, they are removed by apoptosis [10]. Murine HSCs are lineage-negative (Lin−), Sca-1+, and CD117+ (c-kit+) [11]. However, this population contains at least 2 subpopulations with different functions: the first subpopulation consists of cells with a high self-renewal capacity (long-term repopulating HSCs or LT-HSCs) and the second sub-population consists of cells more committed to a particular lineage—short-term repopulating HSCs (ST-HSCs). Mice have very few LT-HSCs in their bone marrow (1–2 cells per 100,000 cells). Exposure of murine bone marrow cells to relatively low doses of radiation resulted in 95% inhibition of the frequency of cobblestone area-forming cells. Also, apoptosis was induced in 65% of Lin−Sca-1+c-kit+ cells. After 5 weeks, 33% of the irradiated cells survived in LT-HSC culture, but could not form myeloid colony-forming units (granulocytes or macrophages). Moreover, the cells that survived expressed an increased level of β-galactosidase and p16, indicating premature senescence [12], perhaps as a protective mechanism to avoid apoptosis during the repair.
Another pathway activated in HSCs after DNA damage is p38 MAPK cascade. It was proved that p38 MAPK pathway is activated by an increase in ROS in murine HSCs. This activation of p38 MAPK limits the life span of HSCs in vivo. In ATM −/− mice, elevation of ROS levels induces HSC-specific phosphorylation of p38 MAPK accompanied by a defect in the maintenance of HSC quiescence [13,14].
The situation in DNA damage control is less clear for mesenchymal SCs (MSCs). It is thought that 2 mechanisms of cellular senescence exist: intrinsic (dependent on telomere shortening) and extrinsic (independent of it). It was demonstrated in the human embryonic lung fibroblast WI38 that busulfan (an alkylation drug that damages DNA mainly through cross-linking) induces senescence in both p53-dependent and -independent pathways and does not induce apoptosis [15]. Similarly Serakinci et al. [16] studied the effect of IR on MSCs isolated from bone marrow. They proved that radiation in doses up to 15 Gy causes growth arrest and senescence but does not induce cell death. Thus MSCs isolated from bone marrow do not die after irradiation by apoptosis but only lose their ability to proliferate, although the molecular mechanism is unknown.
In 2000, Gronthos and coworkers isolated SCs from postnatal human dental pulp (DPSCs) [17]. DPSCs are capable of forming ectopic dentin and associated pulp tissue in vivo. Stromal-like cells were re-established in culture from primary DPSC transplants and re-transplanted into immunocompromised mice to generate dentin pulp-like tissue, demonstrating their self-renewal capability. DPSCs were also found to be capable of differentiating into adipocytes and neuronal-like cells [17,18] and into osteoblasts and endothelial cells [19]. So DPSCs possess SC-like qualities, including self-renewal capability and multilineage differentiation. We isolated DPSCs from deciduous and permanent teeth and cultivated them in a medium with 2% fetal calf serum supplemented with platelet-derived growth factor (PDGF) and epidermal growth factor (EGF). The initial doubling time of our cultures was from 12 to 50 h for the first 40 population doublings [20,21]. The phenotypic analysis show high positivity for CD29, CD44, CD90, and HLA I, while the cells are negative for CD34, CD45, CD71, and HLA II [21].
Radiotherapy is frequently used to treat head and neck cancer. Thus far, nothing is known about the behavior of DPSCs after irradiation. The aim of the present study was to evaluate changes in DPSCs caused by IR in vitro. We studied cell viability, proliferation, apoptosis, and induction of senescence, as well as changes in the expression and phosphorylation status of proteins related to apoptosis and senescence—p53, p21, and p16. The results indicate that senescence does in fact protect these cells from apoptotic death in a manner similar to that observed in HSCs.
Materials and Methods
DPSCs isolation and cultivation
We isolated 4 DPSCs lineages from impacted third molars obtained from healthy donors. The average age of donors was 19 years (12–23, 3 females and 1 male). These or their legal representatives gave written informed consent according to the guidelines of the Ethics Committee of the Medical Faculty in Hradec Králové. Tissue cultures were treated with our standard operation protocols as described previously [20,21]. In briefly, third molars were extracted under sterile conditions and transported to a tissue culture laboratory. After separation of the roots, dental pulp was isolated. Both dental pulp and the tooth were treated by enzymes—collagenase (Sevapharma, Praha, Czech Republic) and dispase (Invitrogen, Carlsbad, CA) for 70 min. Following centrifugation (600g, 5 min), we obtained a cell pellet. DPSCs were cultivated in 5% CO2 atmosphere under 37°C in low FCS cultivation media composed of α-minimum essential medium (MEM; Invitrogen), 2% fetal calf serum (FCS; PAA, Dartmouth, MA), 10 ng/mL EGF (PeproTech, Rocky Hill, NJ), 10 ng/mL PDGF (PeproTech),
Early passages (5th–7th) were then used for the experiments. Cells were seeded at 4,500 cells/cm2 and after reaching 50% confluence they were transported to irradiation facility. Every 4th day following irradiation fresh cultivation medium and growth factors were added and cell passaged as necessary. Cell viability and number of population doublings were examined using Vi-Cell analyzer and Z2-Counter (both from Beckman Coulter, Miami, FL).
γ irradiation
DPSCs in 50% confluence were irradiated at room temperature using a 60Co γ -ray source at a dose rate of 3 Gy/min. After irradiation, flasks were placed in a 37°C incubator and removed at various times following irradiation for analysis.
Apoptosis detection
For apoptosis detection, we used flow cytometric Apoptest-FITC kit (DakoCytomation, Denmark) according to manufacturers' instructions. The kit is based on Annexin V (A) binding to phosphatidylserine at the cell surface of apoptotic cells. Propidium iodide (PI) is used as a marker of cell membrane permeability. Caspase-3 activation was detected using anti-cleaved caspase-3 (Asp175) (Beckman Coulter) according to instructions of the manufacturer. The measurements were performed on Cell Lab Quanta flow cytometer (Beckman Coulter); the data were analyzed using WinMDI 2.9 software.
Cell cycle analysis
In different time of incubation, cells were washed with cold PBS and fixed with 70% ethanol. For detection of low-molecular-weight fragments of DNA, the cells were incubated for 5 min at room temperature in buffer (192 mL 0.2 mol/L Na2HPO4 + 8mL 0.1 mol/L citric acid, pH 7.8) and then stained with PI in Vindelov's solution for 60 min at 37°C. The DNA content was determined by flow cytometer Cytomics FC500 (Beckman Coulter) using 15 mW argon-ion laser with excitation capabilities at 488 nm, and the total emission above 560 nm was recorded. Dual staining cyclin A2/7AAD and p-H3/PI was used according to manufacturers' instructions (Beckman Coulter). The data were analyzed using Multicycle AV software (Phoenix Flow Systems, Inc., San Diego, CA) and CXP (Beckman Coulter).
Reverse transcription and real-time polymerase chain reaction
Total cellular RNA was isolated using RNeasy mini kit (Qiagen, Germany). One microgram of total cellular RNA was transcribed to cDNA with Multiscribe RT enzyme and random primers contained in the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). cDNAs were quantified with TaqMan Gene Expression Assays and TaqMan Universal PCR Master Mix, using the 7500 Fast (Applied Biosystems). The results were normalized to 18S RNA.
Electrophoresis and western blotting
At various times after irradiation, the cells were harvested for preparation of whole-cell lysates (Cell Lysis Buffer, Cell Signaling Technology, Inc., Beverly, MA). The protein content was quantified using bicinchoninic acid (BCA) assay (Sigma-Aldrich, St. Louis, MO). The lysates containing an equal amount of protein (20 μg) were loaded into each lane of a polyacrylamide gel. After electrophoretic separation, the proteins were transferred to a polyvinylidene fluoride (PVDF) membrane (Bio-Rad, Hercules, CA). The membranes were blocked in Tris-buffered saline containing 0.05% Tween 20 and 5% nonfat dry milk and then incubated with primary antibody (p53, p53_serine392—Exbio, Czech Republic; β-actin, p16, p21, mdm2—Sigma-Aldrich; p53_serine15—Calbiochem-Merck, Cambridge, MA; mdm2_serine166—Cell Signaling Technology, Inc.) at 4°C overnight. After washing, the membranes were incubated with appropriate secondary antibody (DakoCytomation, Denmark) for 1 h at room temperature. The signal was developed with a chemiluminiscence detection kit (Roche, Switzerland). To confirm equal protein loading, each membrane was reprobed and reincubated with β-actin.
Histochemistry
DPSCs in 50% confluence were irradiated in chamber slides (NUNC, Denmark). Activity of SA-β-galactosidase was detected by Senescence β-galactosidase Staining Kit (Cell Signaling Technology, Inc.) according to manufacturers' instructions.
Results
Proliferation, viability, and apoptosis of DPSCs after irradiation
Irradiation of DPSCs by the doses of up to 20 Gy had no significant effect on cell viability during the whole interval of the experiment; it varied from about 80% to 90% in the control as well as in the irradiated cells (Fig. 1A). However, IR significantly inhibited proliferation of DPSCs (Fig. 1B and 1C). The total number of cells as well as the cumulative number of population doublings decreased in the dose-dependent manner. Cells irradiated with 2 Gy recovered within 2 weeks and continued with a linear increase of cumulative population doublings later on. The doubling time was also effected—with increased dosages the doubling time increased from 27 h (control) to 128 h (20 Gy, Day 13) (data not shown).
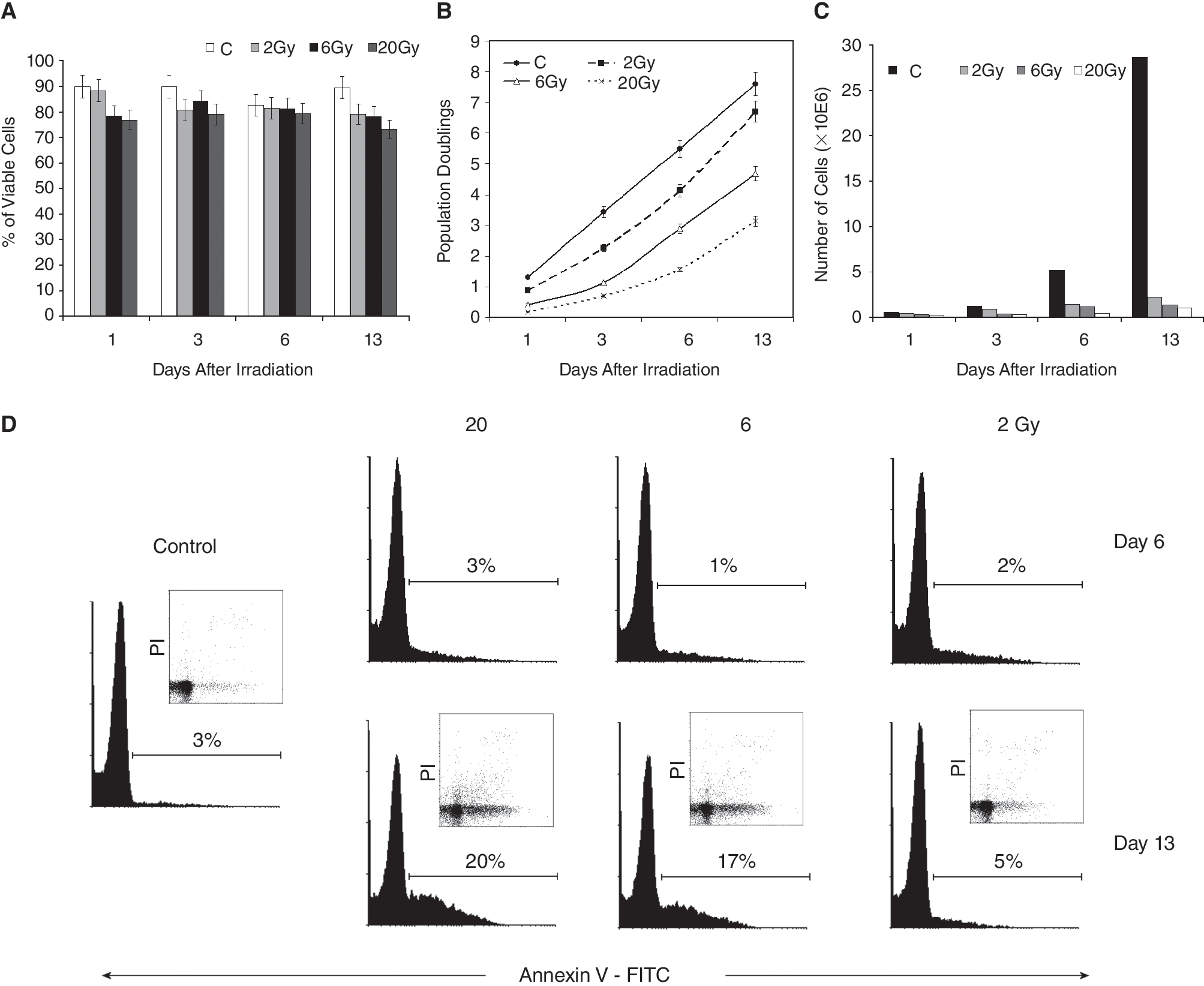
The effect of irradiation of human dental pulp stem cells by the doses of 2, 6, and 20 Gy on the viability, proliferation, and apoptosis during 13 days after irradiation. (
Apoptosis induction occurred in the later interval and was only weak. On Day 13 after irradiation, the percentage of early apoptotic cells (A+/PI−) increased in the dose-dependent manner from 5% (2 Gy) to 17% (6 Gy) and 20% (20 Gy). In the control, 3% of cells were early apoptotic on Day 13 (Fig. 1D). Activation of caspase-3 was not detected (as evaluated by flow cytometry) during the entire experiment (data not shown).
IR induces cell cycle arrest in DPSCs
To determine cell cycle distribution and possible cell cycle arrest after IR, we used cell cycle analysis by flow cytometry. Irradiation with the dose of 2 Gy caused a decrease in the percentage of DPSCs in the S phase of the cycle compared with control cells. The decrease was apparent from the first day after IR (2 Gy 3% vs. control 12%) and this situation persisted throughout the entire experiment (13 days after IR). After doses of 6 as well as 20 Gy, significant cell cycle arrest in the G2/M phase was detected as early as the first day after irradiation (6 Gy 41%; 20 Gy 45%; control 10%). This arrest remained unchanged even on Day 13 post-irradiation (Fig. 2). We confirmed that the cells were arrested in the G2 phase and not in the M phase using PI and phoshorylated histone H3 staining (<1% of cells were detected in the M phase in all groups; data not shown).
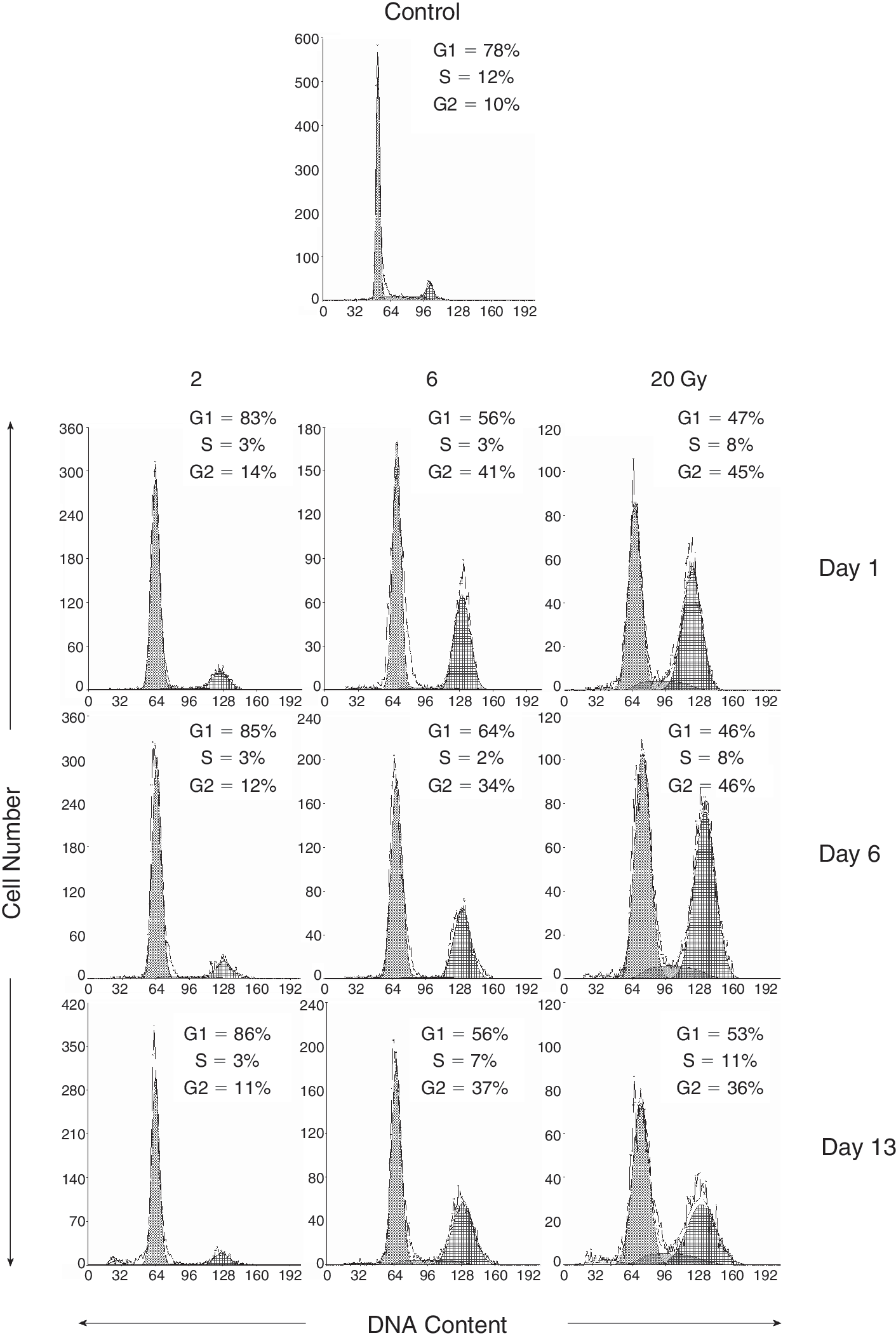
The effects of γ radiation on the distribution of human dental pulp stem cells in the cell cycle and apoptosis induction. Cell cycle distribution and apoptosis were measured using flow cytometric detection of DNA content in the cell. Representative results of cell cycle analysis 1, 6, and 13 days after the irradiation by the doses of 2, 6, and 20 Gy are shown. Significant increase of number of cells in G2/M phase of the cell cycle can be seen after irradiation by the doses of 6 and 20 Gy.
p53 is activated in DPSCs after irradiation
We used electrophoresis and western blotting to study changes in protein expression in human DPSCs after irradiation with a dose of 20 Gy. We observed changes in the level of and phosphorylation status of protein p53 during the 13-day interval. IR caused significant up-regulation of protein p53 in adult DPSCs. Even after only one day of irradiation with a dose of 20 Gy, the level of protein p53 increased and reached its maximum. Three days after irradiation, its level is decreased compared with the previous interval but it was higher than in control. On the 13th day after irradiation, the level of protein p53 dropped back to the level found in control cells (Fig. 3A). Changes in protein p53 were accompanied by its phosphorylation at serine 15, which follows the same tendency. Maximal phosphorylation at serine 15 occurred 1 day after irradiation with a dose of 20 Gy and decreased in time along with the total level of p53. On Day 13 after the irradiation, this phosphorylation was almost undetectable. Phosphorylation of p53 at serine 392 was slower, appearing the first day after irradiation, reaching its maximum 3 days later, and then decreasing in time. On Day 13 after the irradiation, when total protein p53 reached control levels, this phosphorylation was almost undetectable. At the time of p53 elevation, its negative regulator mdm2 is activated via phosphorylation at serine 166 (Fig. 3A).
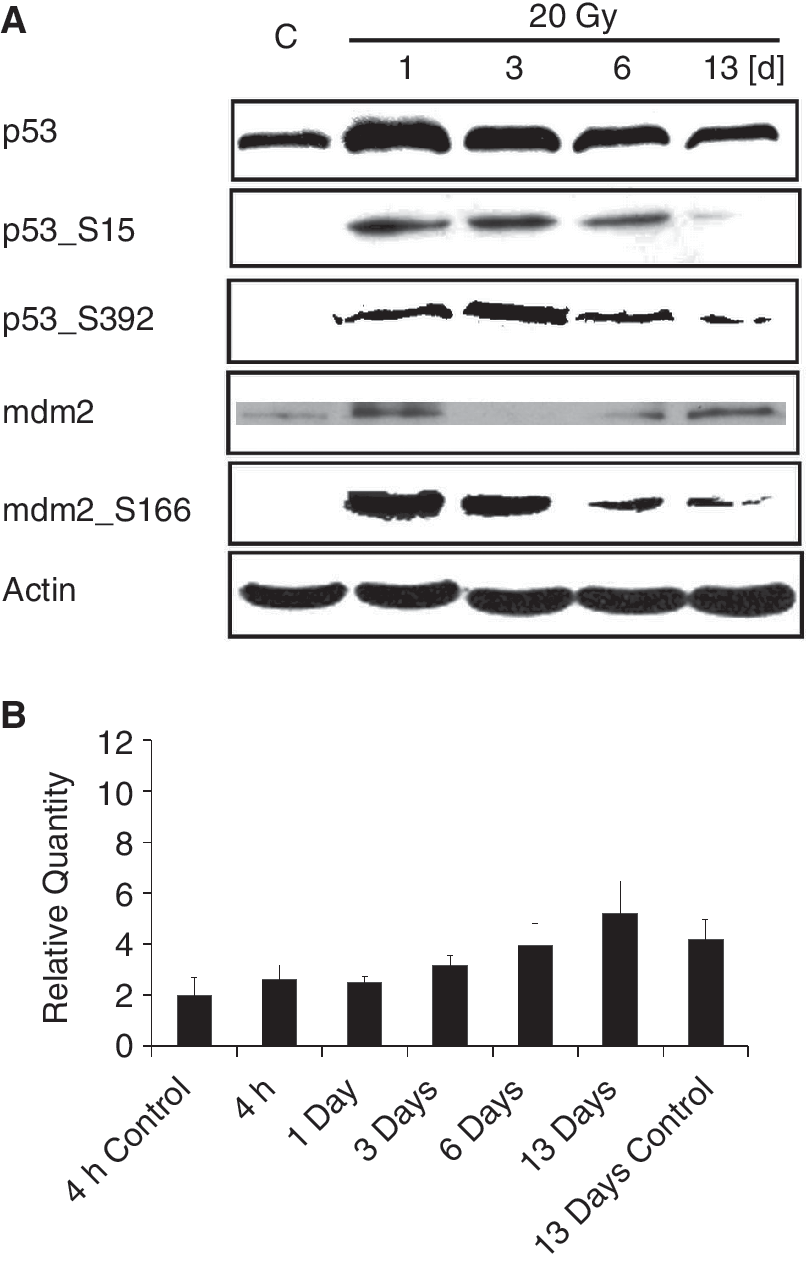
Induction and activation of p53 in human dental pulp stem cells (DPSCs) irradiated with the dose of 20 Gy. (
The increase in the amount of p53 protein was not due to increased expression of TP53 gene, as measured by quantitative reverse transcription polymerase chain reaction (RT-PCR) of p53 mRNA (Fig. 3B).
IR induces p21, p16, and senescence in DPSCs
We studied protein p21 (inhibitor of cyclin-dependent kinase 1A), an inhibitor of cyclin-dependent kinases CDK2 and CDK4. Upon irradiation, the expression of its gene CDKN1A had already increased 4 h after IR, was maximal on Day 1 after the irradiation, and then had diminished in time. By Day 13, it was comparable with control levels (Fig. 4B). The increase in mRNA was accompanied by an increase in corresponding protein p21; a maximal increase in p21 amount (evaluated by electrophoresis and western blotting) was observed on Day 1 after the irradiation and then the p21 amount decreased within the 13 days following irradiation (Fig. 4A).
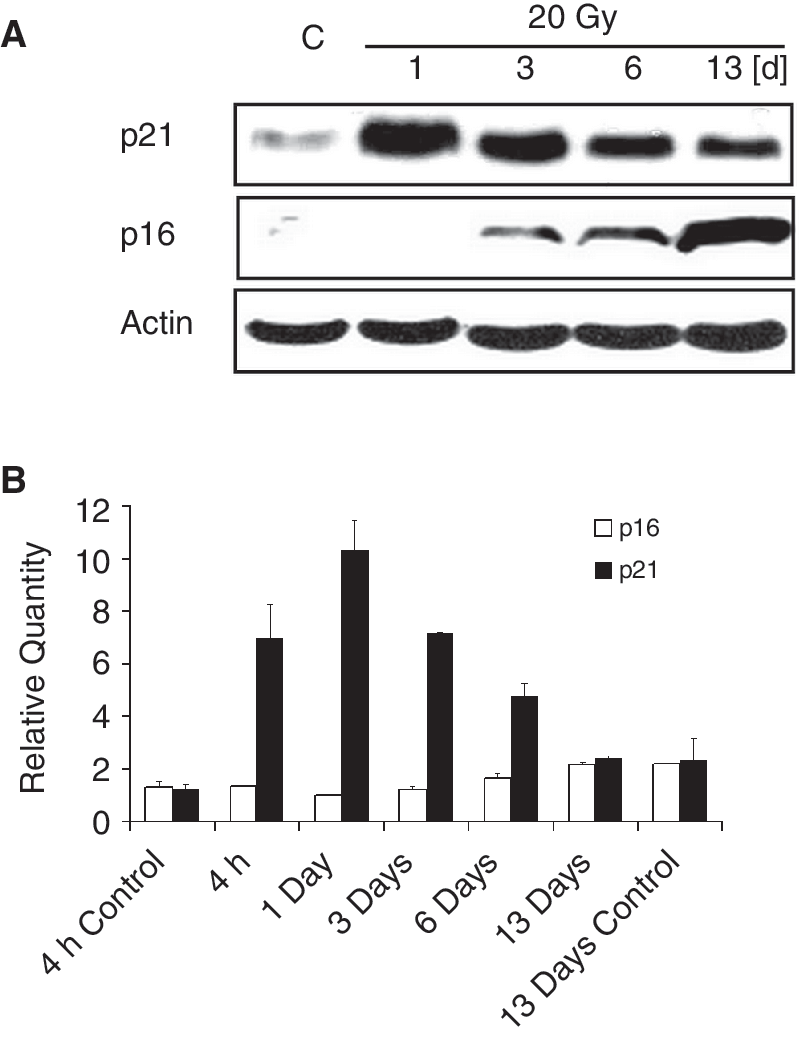
Induction of p21 and p16 in human dental pulp stem cells (DPSCs) irradiated with the dose of 20 Gy. (
The start of the decrease in p21 expression was accompanied by an increase in p16 (inhibitor of cyclin-dependent kinase 2A), a protein that is associated with cellular senescence. This inhibitor of cyclin-dependent kinase CDK4 was first apparent 1 day after irradiation, and its level increased in time for the whole duration of the experiment (13 days). We can conclude that a decrease in p21 levels is followed by an increase in p16 (Fig. 4A). The increase in the amount of p16 protein was not accompanied by an increase in its gene (CDKN2A) transcription, as no increase in p16 mRNA was detected.
Correspondingly, enhanced activity of SA-β-galactosidase can be observed 3 days after irradiation, and its activity increased in time (Fig. 5), indicating the initiation of senescence.
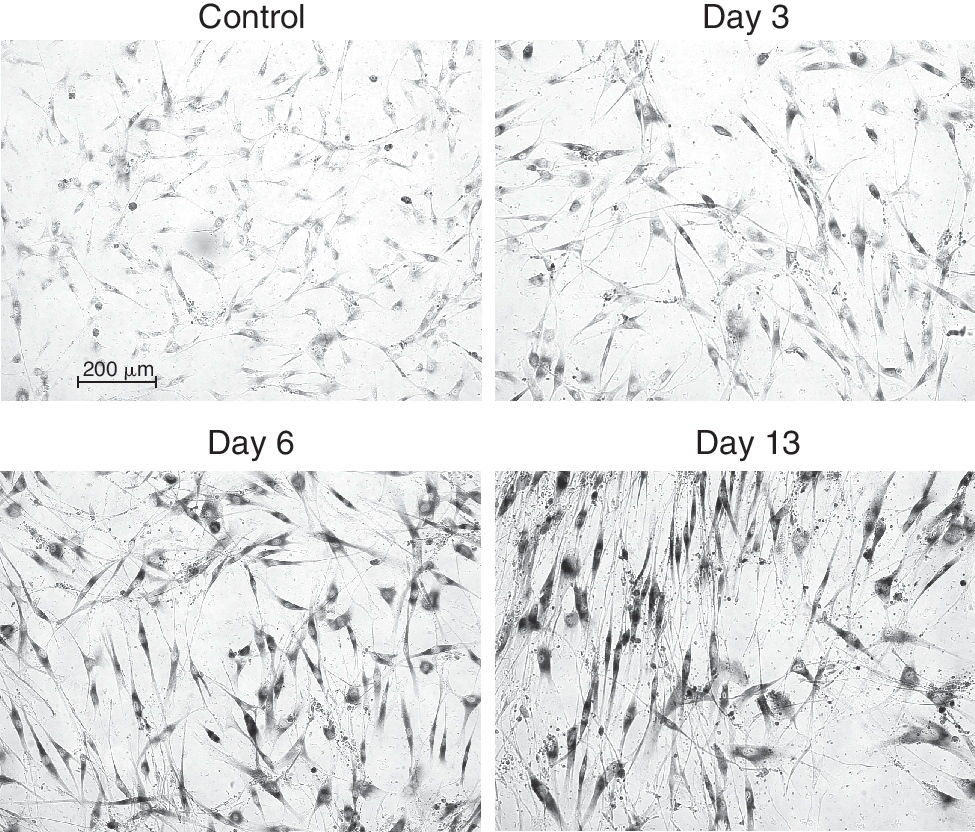
Induction of senescence in human dental pulp stem cells irradiated with the dose of 20 Gy. The cells were irradiated in chamber slides and activity of SA-β-galactosidase was detected histochemically at desired intervals. Activity of SA-β-galactosidase increases from Day 3 after IR, maximal increase was observed at Day 13.
Discussion
Tissue homeostasis depends on the balance among cell proliferation, senescence, and cell death. One of the most important pathways that responds to radiation-induced DSB in eukaryotic cells is phosphorylation of kinase ataxia–telangiectasia-mutated (ATM), and phosphorylation and up-regulation of protein p53 [22,23]. IR-induced damage stops cell cycle progression and triggers temporary cell cycle arrest in the G1/S and G2/M phase. During this period of cell cycle arrest, the cells repair DNA damage, mainly DSB, which are potentially lethal [24,25]. If the repair is unsuccessful, the cells are removed, mostly by apoptosis [26]. However, in some cells the cell cycle arrest becomes permanent. These cells thus lose their proliferative potential, a condition known as premature senescence [27]. Protein p53 is a critically important tumor suppressor protein, which organizes both emergency processes—apoptosis induction and stress-induced premature senescence. The decision as to which of these 2 processes will prevail in the reaction to stress is a multifactorial. It depends on cell type, actual proliferative activity, p53 status, as well as on the ability to up-regulate Cdk inhibitor p16.
In some types of cells, such as fibroblasts, DNA damage (induced, eg, by IR) leads to stress-induced premature senescence. Human fibroblasts react to IR by an increase in p53, p21, and cell cycle arrest in the G1 phase [27 –29], but do not show morphologic hallmarks of apoptosis [29]. The cells in stress-induced premature senescence have all the characteristics of replicative senescence, such as β-galactosidase positivity, p21 accumulation, and hypophosphorylation of pRb. Stress-induced premature senescence is probably induced by p53-dependent cell cycle arrest [27]. The pathway seems to be activated through ATM kinase, phosphorylation of H2AX, formation of γH2AX foci, and loss of telomeric DNA [30].
In this study, we have shown that human adult MSCs isolated from dental pulp react to DNA damage induced by IR mainly by stress-induced premature senescence and not by apoptosis.
We found that these cells lack a G1 checkpoint in response to IR, and are preferentially arrested in the G2 phase of the cell cycle. This mechanism seems to be similar to the response of human embryonic SCs to IR. Filion et al. [31] showed that when human embryonic SCs (ES cells) react to IR, ATM, Ckh2, and p53 are activated and the ES cells are arrested in the G2 phase. They also detected an increase in p21 mRNA levels, but no increase in p21 protein itself.
In our study we found that p53 is up-regulated and phosphorylated on serine 15 and serine 392 in adult DPSCs in response to IR. The phosphorylation of p53 on serine 15 is mainly catalyzed by ATM and is responsible for decreased affinity to ubiquitine-ligase mdm2, thus causing stabilization of p53 [32, 33]. Phosphorylation of serine 392 on the C-terminus of p53 supports its interaction with specific DNA sequences and stabilizes its function as transcription factor [34].
In somatic cells with wild-type p53, including fibroblasts, the activation of p53 results in cell cycle arrest in the G1/S phase transition checkpoint [24,28,29]. The major target gene up-regulated by p53 involved in cell cycle regulation is CDKN1A, a gene encoding protein p21 (inhibitor of cyclin-dependent kinase 1A). p21 acts as an inhibitor of cyclin-dependent kinases CDK2 and CDK4 and in somatic cells causes cell cycle arrest in the G1 phase [35]. We found in our work that DNA damage caused by IR triggers an increase in p53 in DPSCs, followed by an increase in the transcription of the p21 gene CDKN1A, and a significant increase in the amount of protein p21. The increase in p21 peaks during the first 3 days after irradiation. However, this activation of the p53–p21 pathway does not result in G1/S cell cycle arrest, but rather adult DPSCs are arrested in the G2 phase after irradiation. Cell cycle arrest in the G2 phase was apparent within the first day of IR.
A typical response of proliferating cells to massive DNA damage caused by high doses of IR is the induction of apoptosis. However, significant decrease in cell viability and apoptosis were not detected in DPSCs irradiated by doses of up to 20 Gy. In the 20 Gy irradiated DPSCs, the proliferation was inhibited and the cell cycle arrest seemed to be permanent. Instead of apoptosis, we found hallmarks of stress-induced senescence, such as increase in cell cycle regulator protein p16 and increased activity of β-galactosidase, which started to manifest on the third day after irradiation. These data are in good correlation with findings of Serakinci et al., who also detected senescence as a major response of bone marrow MSCs to DNA damage [16]. Our data also correlate with findings on human fibroblasts [28,29].
Our study reveals molecular mechanisms of human adult MSCs isolated from dental pulp in response to DNA damage. In DPSCs, despite the activation of p53 and p21, the cell cycle is arrested in the G2 phase. Apoptosis is not triggered, but p16 increases and the cells enter premature senescence.
Footnotes
Acknowledgments
The work was supported by grant project No. 304/09/1568 of the Czech Science Foundation and grant project NR 91823/07 of the Ministry of Health, Czech Republic.
Author Disclosure Statement
No competing financial interests exist.