
Abstract
Betacellulin (BTC), a ligand of the epidermal growth factor receptor, has been shown to promote growth and differentiation of pancreatic β-cells and to improve glucose metabolism in experimental diabetic rodent models. Mesenchymal stem cells (MSCs) have been already proved to be multipotent. Recent work has attributed to rat and human MSCs the potential to differentiate into insulin-secreting cells. Our goal was to transfect rat MSCs with a plasmid containing BTC cDNA to guide MSC differentiation into insulin-producing cells. Prior to induction of cell MSC transfection, MSCs were characterized by flow cytometry and the ability to in vitro differentiate into mesoderm cell types was evaluated. After rat MSC characterization, these cells were electroporated with a plasmid containing BTC cDNA. Transfected cells were cultivated in Dulbecco's modified Eagle medium high glucose (H-DMEM) with 10 mM nicotinamide. Then, the capability of MSC-BTC to produce insulin in vitro and in vivo was evaluated. It was possible to demonstrate by radioimmunoassay analysis that 104 MSC-BTC cells produced up to 0.4 ng/mL of insulin, whereas MSCs transfected with the empty vector (negative control) produced no detectable insulin levels. Moreover, MSC-BTC were positive for insulin in immunohistochemistry assay. In parallel, the expression of pancreatic marker genes was demonstrated by molecular analysis of MSC-BTC. Further, when MSC-BTC were transplanted to streptozotocin diabetic rats, BTC-transfected cells ameliorated hyperglycemia from over 500 to about 200 mg/dL at 35 days post-cell transplantation. In this way, our results clearly demonstrate that BTC overabundance enhances glucose-induced insulin secretion in MSCs in vitro as well as in vivo.
Introduction
D
Mesenchymal stem cells (MSCs) can be isolated from bone marrow based on their ability to adhere to plastic substrates [12]. Under appropriate conditions, they differentiate into multiple mesenchymal cell types including cartilage, bone, adipose and fibrous tissues, and myelosupportive stroma [13]. Recent work has attributed to MSCs the potential to differentiate into cells of all 3 primary germ layers, including mesoderm, neuroectoderm, and endoderm; they are also reported to be immunoprivileged and immunosuppressive [11]. Rat [14] and human MSCs [15] have been shown to differentiate into insulin-secreting cells.
Davani et al. [16] have shown that human islet-derived precursor cells are capable of differentiating to adipocyte, condrocyte, and osteocyte, human islet-derived precursor cells also express MSC markers, indicating that these cells could be MSCs that are present in adult islets in situ. Betacellulin (BTC) is a multifunctional polypeptide growth factor belonging to the family of epidermal growth factor receptor (EGFR) ligands [17]. BTC was first described as a mitogen from a mouse pancreatic insulinoma cell line [18]. BTC is initially produced as a transmembrane protein that can be cleaved by metalloproteinase to release the mature circulating form [19]. Regarding the tissue distribution, BTC is expressed abundantly in the pancreas and intestine [20]. There are several evidences indicating that, among the 7 EGFR ligands, BTC exerts unique actions in pancreatic islet physiology. BTC converts pancreatic progenitors to insulin-secreting cells in vitro [21 –23], sustains PDX1 expression, and induces β-cell differentiation in human ES cells [24].
In addition, the administration of this growth factor in vivo improves glucose metabolism by increasing the β-cell mass in diabetic animal models [22,25]. These results suggest that BTC plays an important role in regulating growth and differentiation of pancreatic endocrine precursor cells.
Considering the plasticity of MSCs and the action of BTC in differentiation and proliferation of β-pancreatic cells, our aim was to evaluate the in vitro and in vivo effects of BTC overexpression in rat MSCs.
Materials and Methods
Transgene construction
The BTC-coding sequence was amplified from mouse lung cDNA by polymerase chain reaction (PCR) using the primers: BTC 5′-GGC CCA GGA AGG GCA TAG AGA-3′ and BTC 5′-ATG AGT CAG GTC TTT TGT AGC TTG-3′ [26]. The product was inserted into the pCRII-TOPO cloning vector (Invitrogen), and after sequencing, cDNA was cloned into EcoRI site downstream of the cytomegalovirus promoter and upstream to the internal ribosome entry site (IRES) and the enhanced green fluorescent protein (EGFP) in the expression vector pIRES2-EGFP (Clontech). Correct orientation was checked by digestion with KpnI and SmaI. The constructed vector was named pIRES2-BTC-EGFP.
Isolation and culture of bone marrow cells
Eight-week-old Wistar rats were purchased from the Centro de Reproduçáo e Experimentaçáo de Animais de Laboratório–CREAL-UFRGS. The procedures were performed in accordance with the guidelines for animal experimentation of UFRGS University. Bone marrow cells were obtained from femurs and tibias. After isolation, 1 × 107 bone marrow-derived cells were cultured (37°C, 5% CO2) in T25 culture flasks (TPP) with DMEM (Invitrogen) medium containing 15 mM HEPES, 15% inactivated fetal bovine serum (FBS; Invitrogen), 100 U/mL penicillin, and 100 mg/mL streptomycin antibiotic solution (Gibco). On the third day of culture, medium was changed and nonadherent cells were removed. Adherent cells gaining 80% of confluence were passaged with the use of 0.05% trypsin–EDTA solution (Gibco) and then maintained in Dulbecco's modified Eagle medium (DMEM) with 10% FBS (complete medium).
Differentiation assays
To characterize MSCs in accordance with the International Society for Cellular Therapy statement [27], 2 different experimental procedures were employed. Osteogenic differentiation was induced by culturing MSCs for up to 3 weeks in DMEM containing 10% FBS and 15 mM HEPES, supplemented with 10−8 M/L dexamethasone (Sigma), 5 μg/mL ascorbic acid 2-phosphate (Sigma), and 10 mM/L β-glycerolphosphate (Sigma). To observe calcium deposition, cultures were stained with alizarin red S stain (Nuclear).
Second, to induce adipogenic differentiation, MSCs were cultured with 10−8 M dexamethasone (Sigma), 5 μg/mL insulin, and 50 μg/mL indomethacin (Sigma). Adipocytes were easily discerned from the undifferentiated cells by phase-contrast microscopy. To further confirm their identity, cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) and stained with oil red (Sigma) on day 21 of adipogenic differentiation.
Flow cytometry
Approximately 1 × 106 MSCs were prepared. They were placed in sterile tubes and washed 2 times by centrifugation at 300 g for 5 min at 4°C. MSCs were then resuspended in PBS and incubated for 20 min at 4°C with phycoerythrin- or fluorescein isothiocyanate-conjugated antibodies against rat CD34, CD45, CD11bc, CD44, CD90, and CD29. All assays were conduced using antibody concentrations as recommended by the manufacturers. Phycoerythrin and fluorescein isothiocyanate mouse anti-rat IgG1, IgG2a, and IgM were used as isotype controls. Cells were collected and washed with PBS by centrifugation and fluorescence analysis was carried out with the BD FACS-Calibur flow cytometry system (Becton-Dickinson) with a one-laser system that is capable to detect 3 fluorochromes excited by the 488 nm laser in a multiparameter manner. Data samples were analyzed using Cellquest and PAINT-A-GATE software.
Cell transfection
MSCs were harvested by trypsinization and then pelleted via centrifugation. Excess media were removed to obtain a tight cell pellet, which was resuspended in 4°C DMEM without serum or antibiotics. Cells were quantified in Neubauer's chamber and 1 × 106 cells in pure DMEM were transferred to a 4-mm cuvette with 60 μg of pIRES2-BTC-EGFP or pIRES2-EGFP (empty vector used as negative control). After an incubation time of 5 min at room temperature, cells were electroporated using Gene Pulser XCell (Bio-Rad Laboratories) by applying the following parameters: 950 μF, 200 Ωm, and 350 V. After electroporation, cells were incubated for 5 min at room temperature and plated in H-DMEM complete medium supplemented with 10 mM nicotinamide (Acros). Twenty-four hours after electroporation, medium was replaced and cells were analyzed under fluorescence microscopy. After 48 h, cells were submitted to the selection media using G418 (Geneticin; Gibco).
Immunocytochemistry assay
For BTC immunofluorescence, pIRES2-BTC-EGFP–transfected cells were fixed with 4% formaldehyde in PBS. The primary antibody, goat anti-mouse BTC (R&D Systems), was diluted 1:100 and incubated overnight at 4°C. A Cy3-conjugated rabbit anti-goat secondary antibody (Sigma) was used. Fluorescence was detected using fluorescence microscopy (Nikon Eclipse TE 2000-U; Nikon Instruments).
For insulin immunocytochemistry assay, pIRES2-BTC– and pIRES-EGFP–transfected cells were fixed with 4% formaldehyde in PBS. We used a 1:500 dilution of guinea pig anti-insulin (Dako) and Universal Dako LSAB Kit Peroxidase (Dako). Cells were analyzed under microscopy (Nikon Eclipse TE 2000-U; Nikon Instruments). The same antibody was used for immunohistochemistry of the grafts.
Insulin radioimmunoassay
The static incubation method was applied as described by Lumelsky et al. [28].
Briefly, 105 adherent cells at 4, 8, 12, and 16 days posttransfection were rinsed twice with Krebs-Ringer with bicarbonate buffer (120 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 1.1 mM MgCl2, 25 mM NaHCO3, and 0.1% bovine serum albumin). An inhibitor of insulin secretion (Nifedipine; Sigma) was added to the medium at 30 min prior to medium replacement with Krebs-Ringer containing 2.7 mM glucose. Cells were incubated for 15 min at 37°C with Krebs-Ringer and then in buffer containing 5, 30, or 50 mM glucose for 1 h.
Insulin levels in cell supernatants were measured using an immunoradiometric assay kit (RIA 13k; Linco Research). To each polypropylene tube, anti-insulin, 125I-Insulin, and 100 pL of the samples were added. Immune complexes were precipitated 24 h later with polyethylene glycol solution, and a gamma counter was used to determine the radioactivity in the precipitates. Four determinations were carried out in duplicate and the means and standard error were obtained.
RNA extraction and reverse transcription–PCR
Reverse transcription (RT)–PCR was performed to assess the expression of pancreatic β-cell–related genes (PDX1, PAX4, INS1, and NKX6.1).
Total RNA was isolated from nontransfected MSCs, MSCs at 14 days after transfection with pIRES2-EGFP-BTC or pIRES2-EGFP, and RINm5f cell line (used as a positive control for pancreatic genes expression). Total RNA was isolated using the SS III First Strand super mix (Invitrogen). Standard RT was performed using M-MLV Reverse Transcriptase (Sigma) according to the manufacturer's instructions. Reactions mixtures for PCR included 1 μg of cDNA, 0.2 μM of each antisense and sense primer, 0.2 mM dNTPs, 1.5 mM MgCl2, and 1 U of Taq DNA polymerase (Invitrogen). PCR was performed on a thermocycler Techne TC412 (Barloworld Scientific). Amplification conditions consisted of an initial denaturation step at 94°C for 1 min, followed by 40 cycles of denaturation at 94°C for 30 s, annealing for 1 min, extension at 72°C for 30 s, and final polymerization at 72°C for 10 min. β-Tubulin was used as an internal standard.
Diabetes animal model
Male Wistar rats housed at 22°C in an air-conditioned environment, with a 12-h light–dark cycle, and fed a regular unrestricted diet received a single intraperitonial injection of 70 mg/kg streptozotocin (Sigma) freshly dissolved in 0.1 M citrate buffer (pH 4.5).
Blood glucose was measured weekly with a glucometer (Optium Xceed; Abbott). Animals whose blood glucose level exceeded 300 mg/dL were considered diabetic.
Cell transplantation
Under anesthesia, a medial abdominal laparotomy was performed and the left kidney was exposed. About 4 × 106 cells in 50 μL of PBS were transplanted into the kidney parenchyma by direct insertion of a 24-gauge syringe into the kidney capsule at the upper pole and cells were gently discharged into the parenchyma. Blood glucose levels were monitored once a week (days 14, 21, 28, and 35) in samples obtained from the tail vein, using a glucometer.
At the end of the experiment (day 35), rats were euthanatized and kidneys and pancreas removed for hematoxylin and eosin staining for histological analysis.
Statistical analysis
Data are expressed as mean ± SE and statistical differences between groups were determined by repetitive measure ANOVA in SPSS version 16. Bonferroni post-hoc test was applied when necessary. Differences between groups were considered significant when P < 0.05.
Results
Phenotypic characteristics of expanded undifferentiated MSCs
Bone marrow MSCs were obtained from rats by plating bone marrow cell suspension in tissue culture dishes and propagation of adherent cells. The isolated cells developed into visible systematic colonies of adherent fibroblast-like cells at about 7–10 days after initial plating and became morphologically more homogeneous (Fig. 1A, B) with time in culture by depletion of hematopoietic and other bone marrow stromal cells. Rat MSCs are known to be positive for CD44, CD90, and CD29 and negative for CD34, CD45, and CD11bc. Flow cytometry was performed from the moment of MSC extraction to the third trypsinization step. By the third passage, the vast majority of cells stained for the markers CD44 (99.3%), CD90 (99.8%), and CD29 (99.5%) and only a small proportion manifested expression of the markers CD45 (0.9%), CD11b/c (0.52%), and CD34 (0.05%). According to this pattern of cell surface marker expression, the cell population at this time point (∼24 days in culture) was quite uniform and can be considered bona fide MSCs.
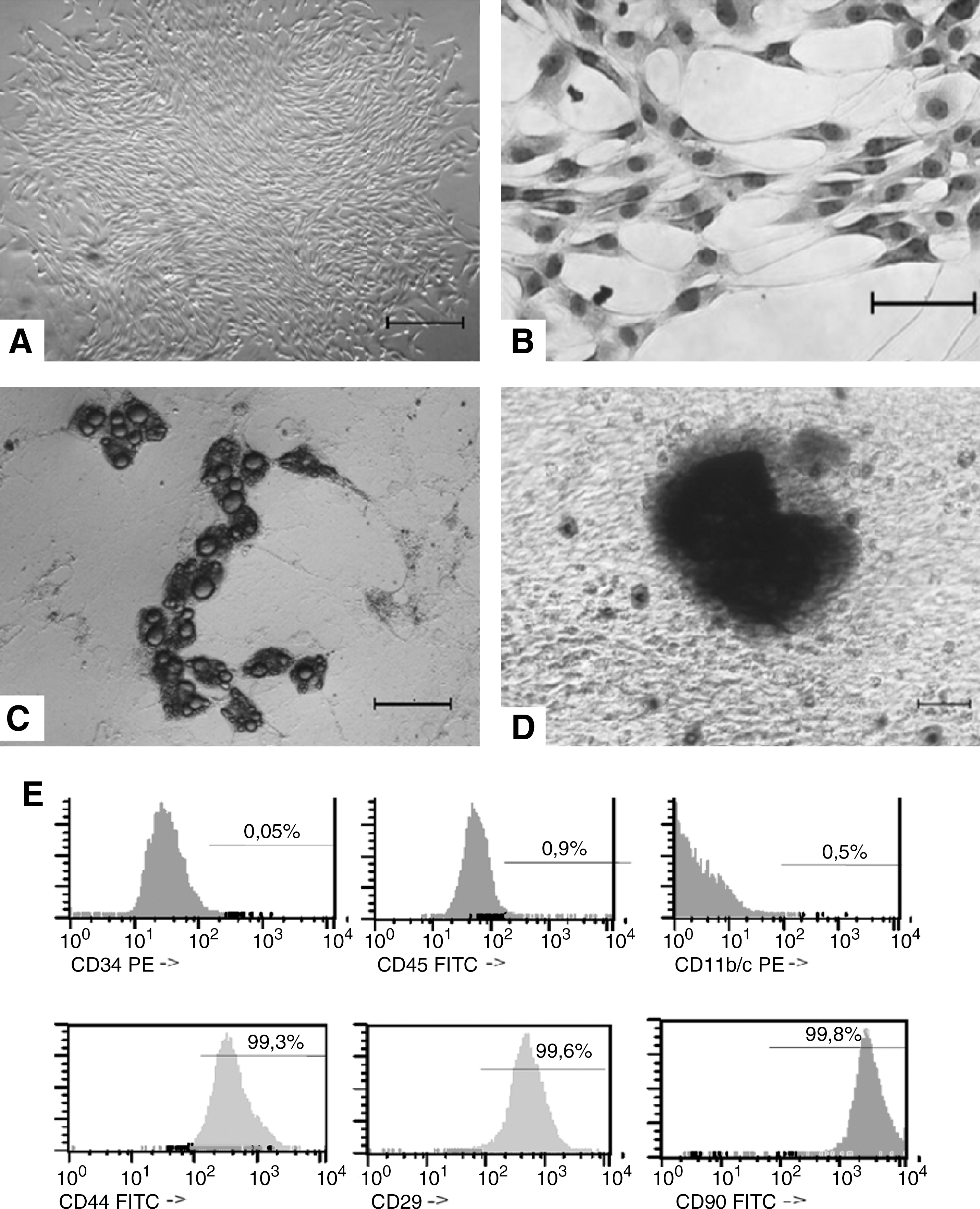
Characterization of MSCs.
The MSC differentiation potency was shown using protocols known to induce differentiation into bone and adipose cells. The results showed a clear potency for adipogenic differentiation as detected by oil red staining and for osteogenic differentiation as detected by deposit of calcium (Fig. 1C, D, respectively).
BTC expression in vitro modified the morphology and gene expression pattern in MSCs. After characterization, MSCs within 12 passages were transfected by electroporation with the expression vector pIRES2-BTC-EGFP or the empty vector. The vector included neomycin resistance gene and an IRES sequence of the encephalomyocarditis virus in the multiple cloning site, permitting both BTC and EGFP cDNAs to be translated from a bicistronic mRNA.
The efficiency of transfection with this vector as evaluated by the number of GFP-positive cells was ∼40%. Following selection, the surviving cells (termed BTC-MSCs or pIRES-MSCs, depending on the vector employed) were analyzed.
BTC-MSCs showed GFP expression and immunofluorescence for BTC (Fig. 2). No expression of BTC was detected in MSCs and pIRES-MSCs (data not shown).
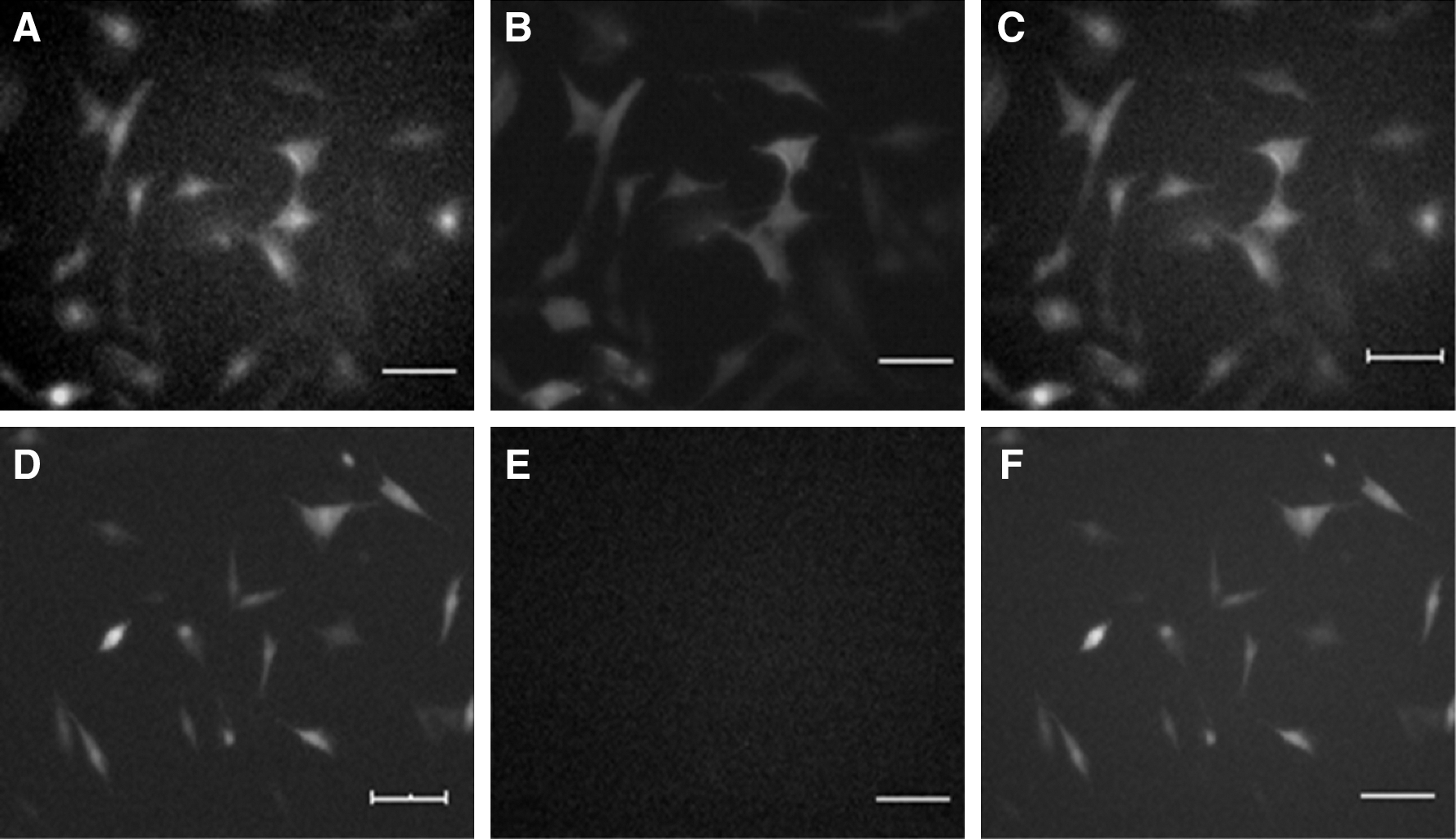
Expression of BTC and GFP in BTC-MSCs and pIRES-MSCs.
Under inversed microscope, it was possible to observe within 10 days that the morphology of BTC-MSCs changed from spindle-like (Fig. 3B) to flat epithelial-like cells (Fig. 3A).
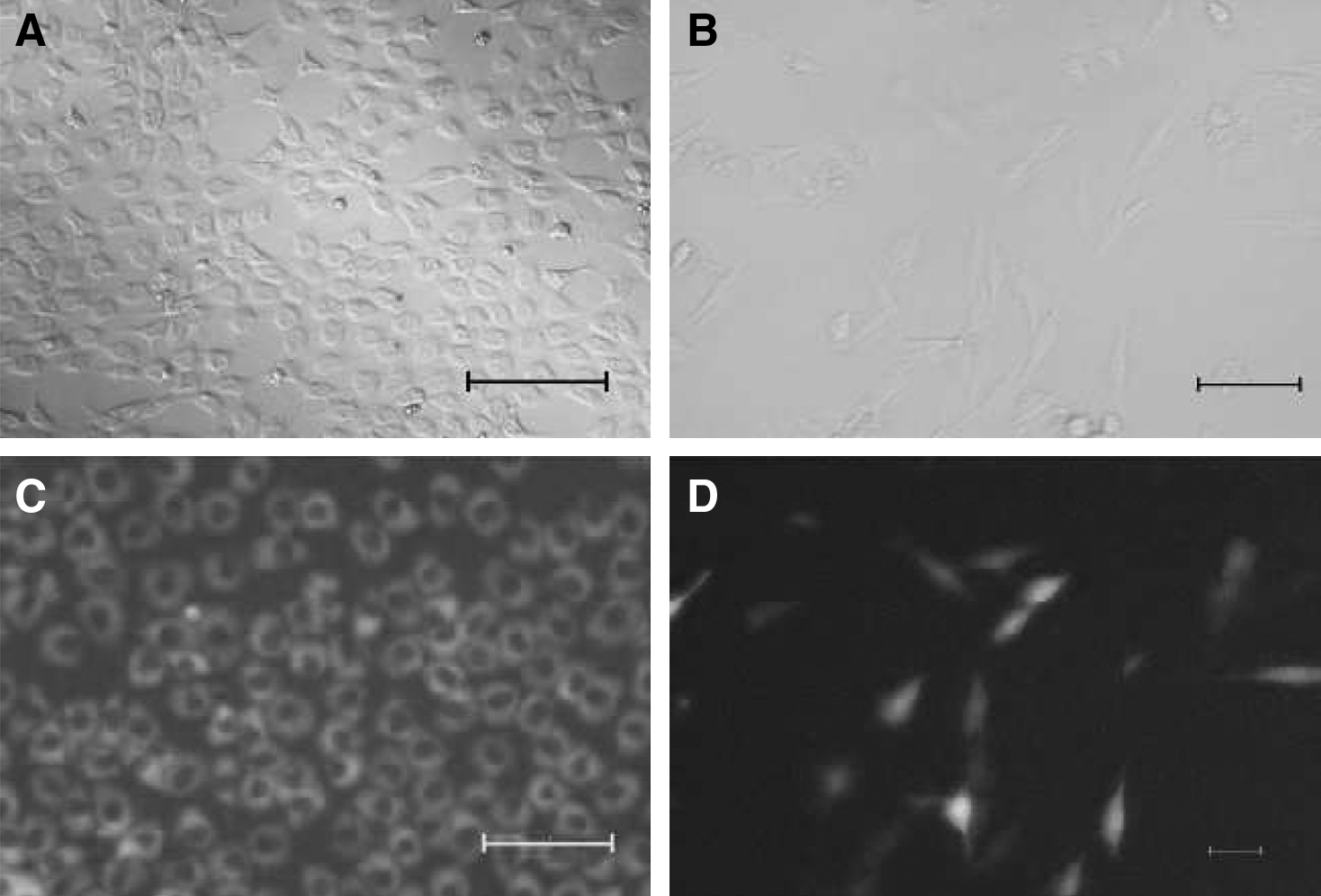
Morphologic analysis of BTC-MSCs and pIRES-MSCs at 10 days after transfection.
To assess the mRNA expression of typical β-cell genes (PDX1, PAX4, INS1, and NKX6.1), RT-PCR was applied on RINm5f insulinoma cells (used as positive control for pancreatic gene expression), MSCs (negative control), pIRES-MSCs, and BTC-MSCs. Transcripts of PDX1, PAX4, INS1, and NKX6.1 were detected at 14 days after transfection in MSC-BTC but not in MSCs or MSC-pIRES, as expected (Fig. 4). The results strongly suggest that the overexpression of BTC may induce the expression of multiple islet genes involved in the β-cell differentiation pathway.
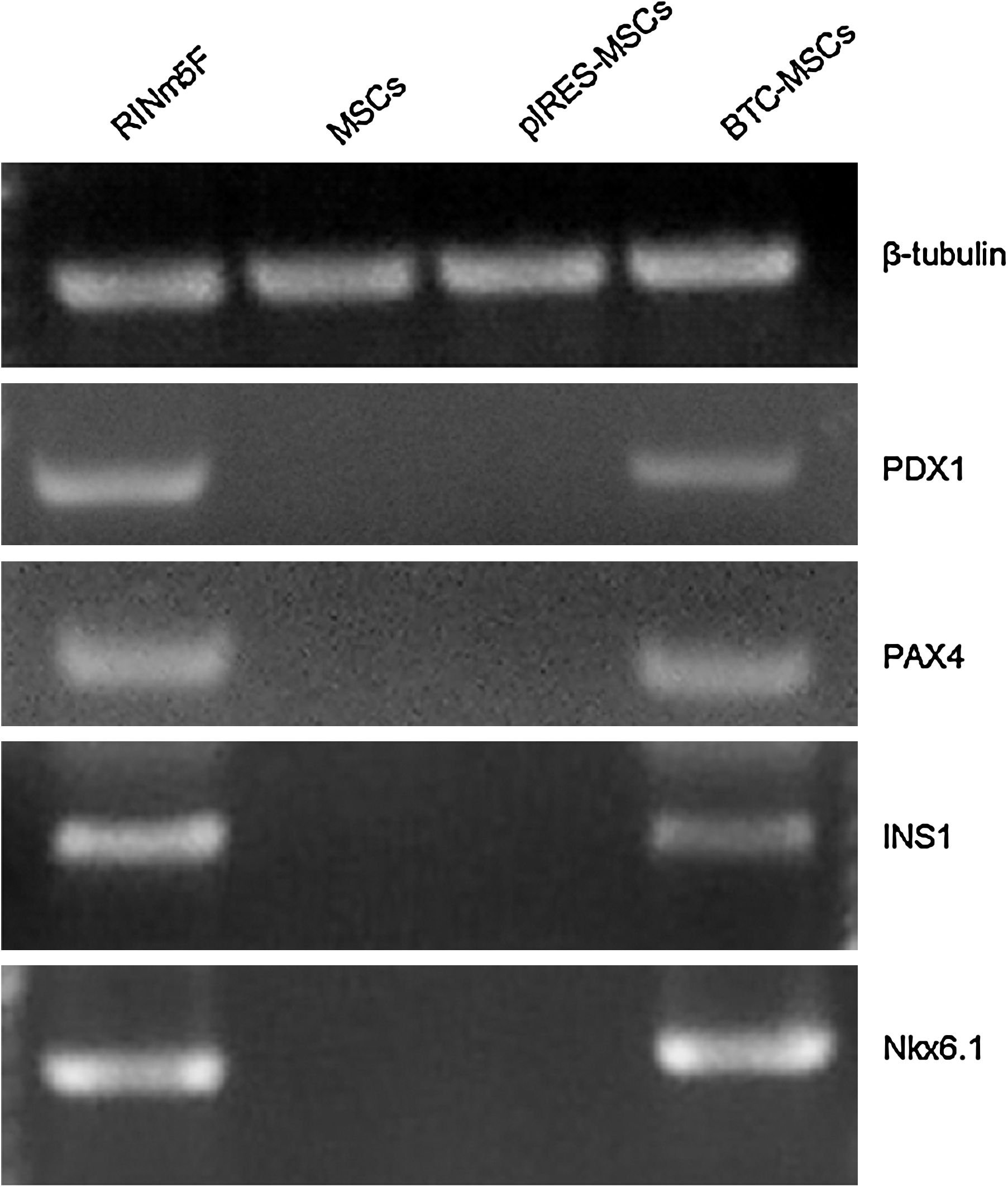
Reverse transcription–polymerase chain reaction analysis of mRNA expression of the indicated genes in RINm5F, MSCs, pIRES-MSCs, and BTC-MSCs.
After 23 days of culture, different from pIRES-MSCs, some BTC-MSCs presented aggregating tendency and formed a 3D structure (Fig. 5A, B) similar to the structures described by other authors during specific differentiation protocols [29 –31].
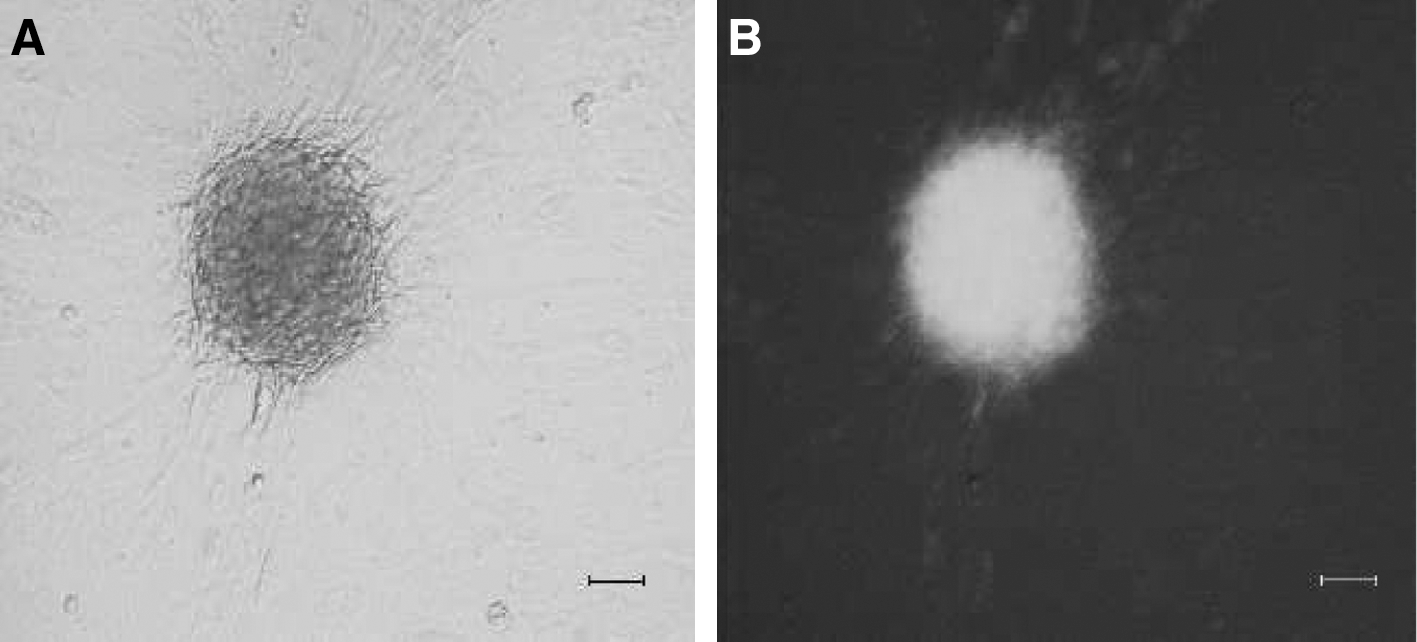
BTC-MSCs tended to aggregate and formed a 3D structure similar to islet-like spheroids.
MSC-BTC cells develop insulin expression and secretion
To evaluate the functional differentiation of MSC-BTC, they were tested for insulin content and glucose-induced insulin secretion in vitro.
Immunocytochemistry analysis demonstrated positive staining for insulin (data not shown) in BTC-MSCs with the epithelial-like morphology after 12 days of culture. As expected, we could not detect positive stain for insulin in pIRES-MSCs.
To assess the cell's ability to produce and secrete insulin, RIA was used to measure insulin secretion from BTC-MSCs and pIRES-MSCs at different time points: 4, 8, 12, and 16 days after transfection. In vitro insulin secretion from BTC-MSCs was modulated by glucose and was first detected on day 8 and continued until day 16 posttransfection (Fig. 6). In contrast, in pIRES-MSCs, insignificant immunoreactive insulin was detected at all time points (P = 0.01).
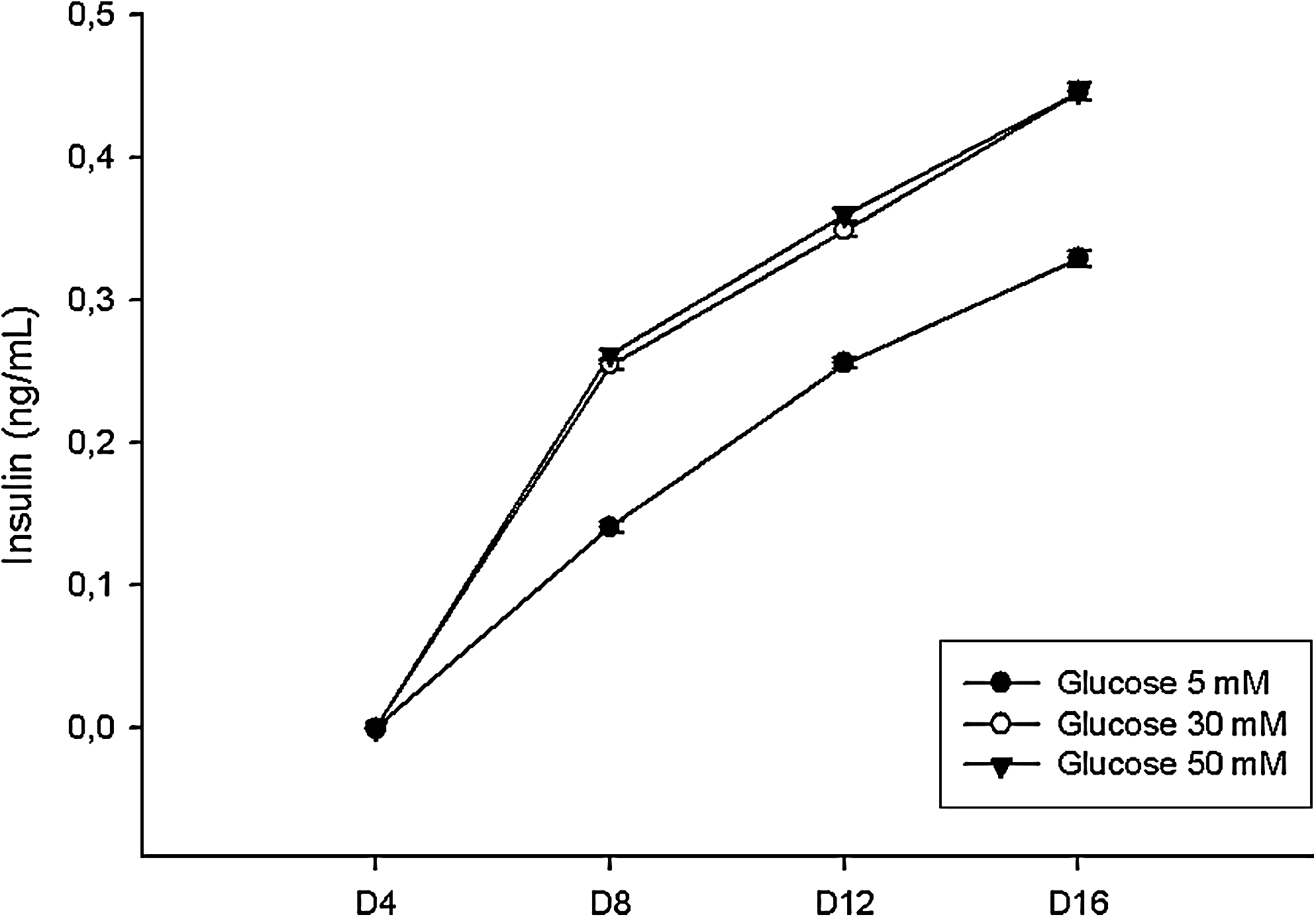
In vitro glucose-induced insulin secretion in BTC-MSCs. The amount of insulin in the supernatant was determined by radioimmunoassay.
Transplantation of BTC-MSCs reverts hyperglycemia in streptozotocin diabetic rats. Transplantation of BTC-MSCs under the renal capsule of streptozotocin diabetic rats was able to modulate glycemia in vivo as confirmed by the reduction of blood glucose levels, beginning 14 days posttransplantation from over 500 to about 200 mg/dL at 35 days posttransplantation (P = 0.001) (Fig. 7A). In contrast, in streptozotocin (STZ) diabetic rats transplanted with control pIRES-MSCs, blood glucose levels continued to be high.
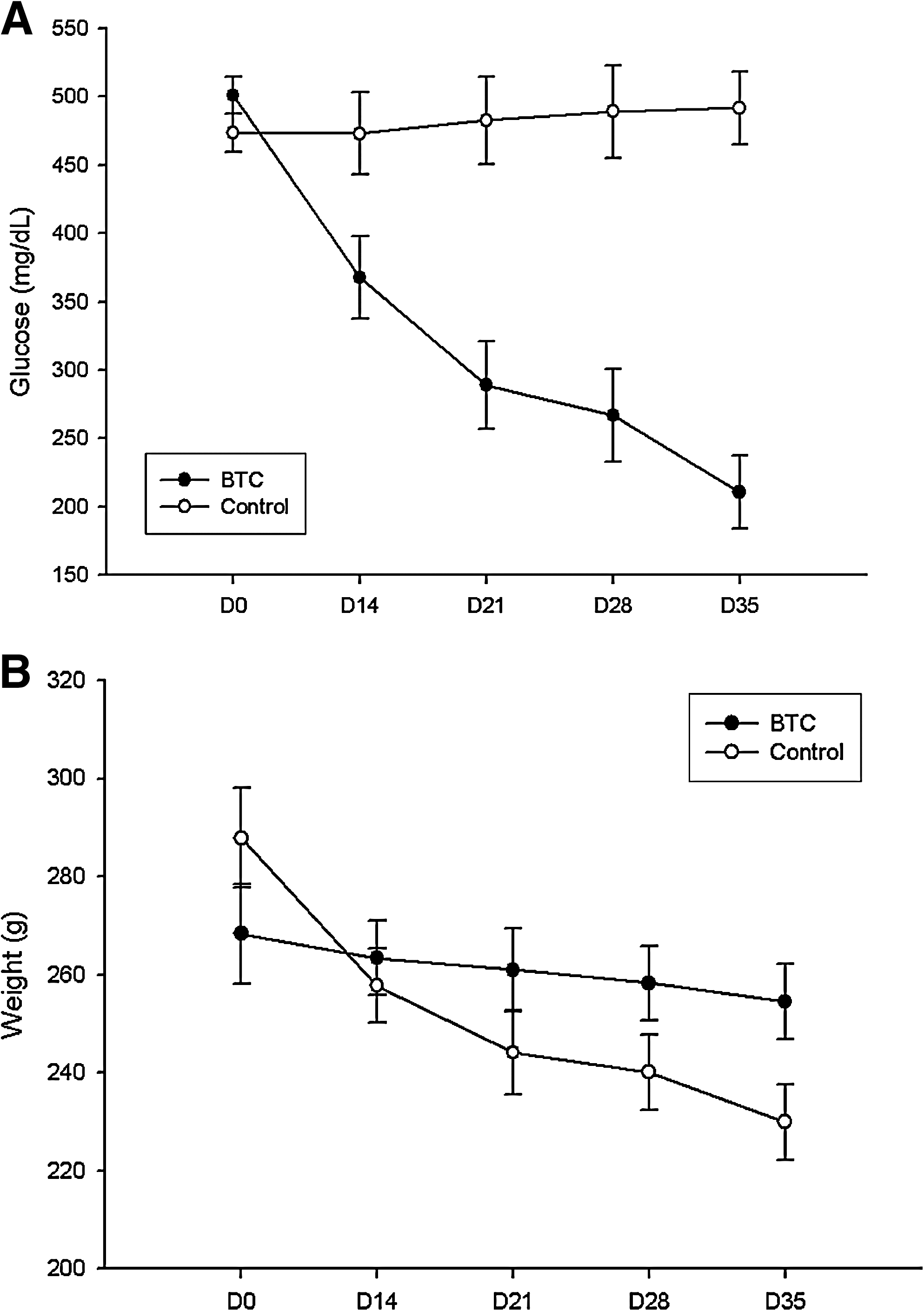
Functional analysis of BTC-MSCs and pIRES-MSCs in vivo.
In parallel, streptozotocin (STZ) diabetic animals that received BTC-MSCs showed a less-severe weight loss (Fig. 7B) when compared with the pIRES-MSC animals (P = 0.01).
No gross tumor formation was seen in the animals and no tumor-like structures were observed among the grafts by histological analysis (Fig. 8). It was possible to observe the presence of insulin-positive cells in the renal subcapsular area of BTC-MSC–transplanted animals (Fig. 9).
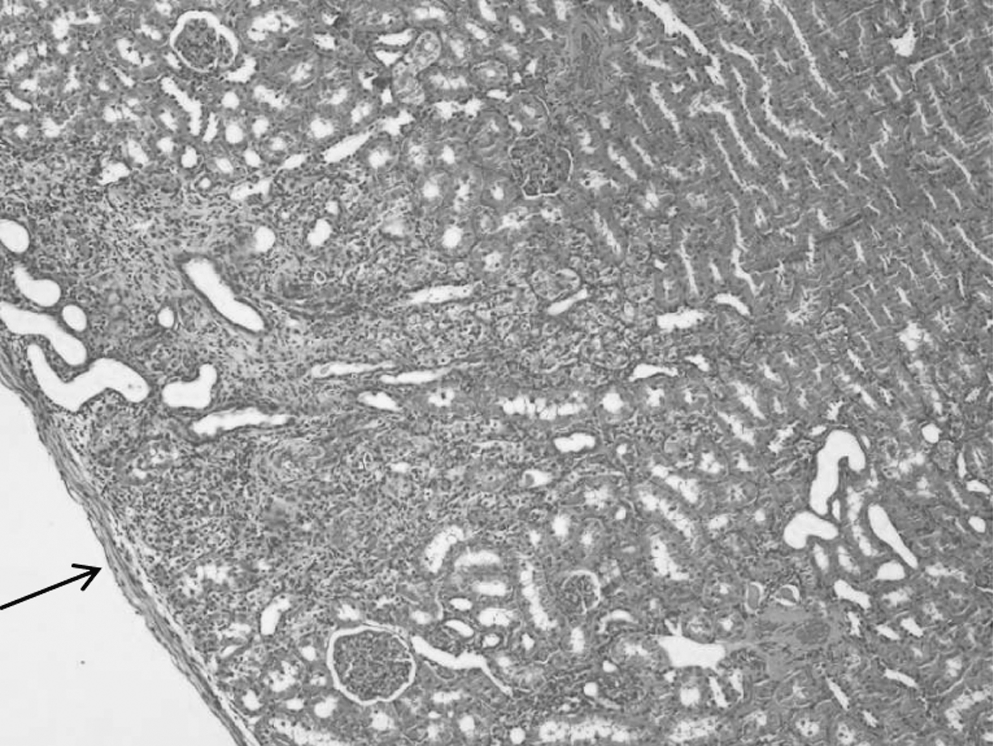
Histological analysis of the graft area. Hematoxylin and eosin staining showing the presence of transplanted cells under the renal capsule (200 ×). Arrow indicates the graft area.
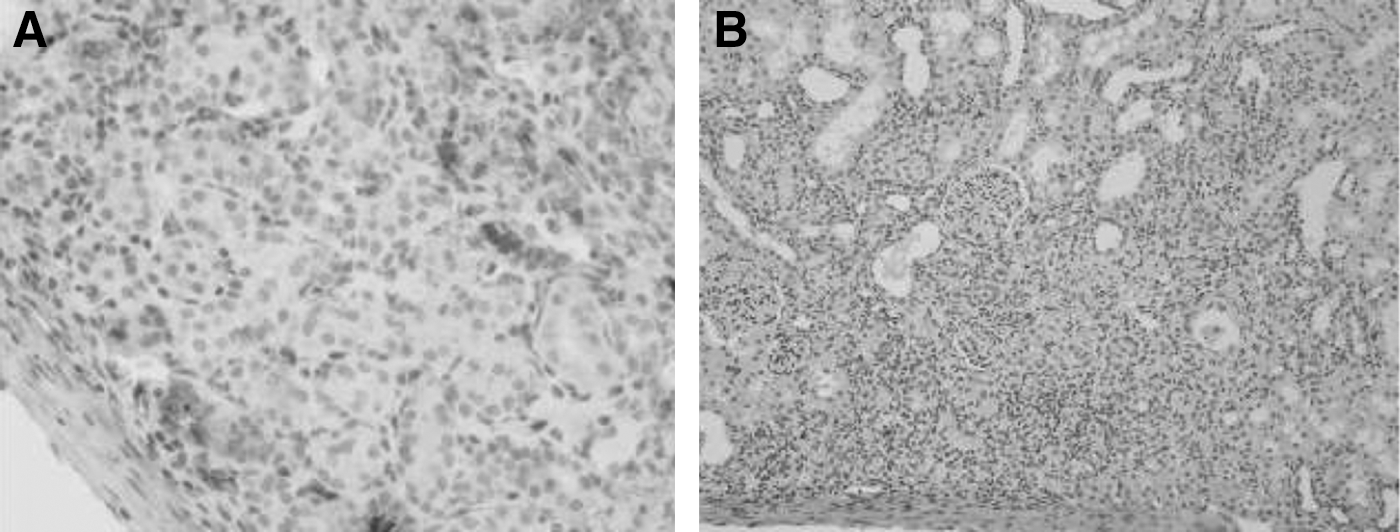
Immunohistochemistry analysis of the transplanted kidney.
Discussion
Although pharmacological therapy for diabetes has continued to improve, tight glucose control has not eliminated the complications of diabetes. Pancreas or islet transplantation can reconstitute the glucose-regulatory function of normal islets but is limited by an inadequate donor supply, need for immunosuppression, and loss of function of the transplanted islets [1].
In this regard, sources of pancreatic β-cells other than those from organ donors are needed. In the present study, we have devised a strategy to induce the formation of insulin-producing cells by overexpressing BTC in rat MSCs cultivated with nicotinamide. Previous studies have provided evidences that there are 2 pathways of pancreatic regeneration: (a) replication of pre-existing differentiated exocrine and endocrine cells and (b) proliferation and subsequent differentiation of duct epithelium to form new islets [32]. BTC is involved in both pathways; it is known to induce proliferation of insulinoma cells in vitro [33], improve glucose tolerance, enhance insulin secretion [34], reverse streptozotocin-induced hyperglycemia in mice [35], and induce endocrine differentiation of the exocrine cell line AR42J [23]. This differentiation potency can be explained because EGFR activation affects cell function on multiple levels, depending on the signaling pathways that are activated [18]. Our in vitro results indicate that the overexpression of BTC in rat MSCs may induce the expression of PDX1 as well as other important pancreas transcription factors, and the consequent production of insulin in agreement with Moriscot's conclusions about MSCs [36]. We observed that the morphology of BTC-MSCs changed from spindle-like to flat epithelial-like cells. This morphologic differentiation could be related to the findings of Gershengorn et al. [37] and Russ et al. [38], who believe in the theory of epithelial-to-mesenchymal transition where cell morphology differentiation is related to changes in gene expression from mesenchymal to epithelial patterns and vice versa.
In this way, others have observed similar morphologic differentiation trying to obtain endocrine cells from MSCs and attributed it to mesenchymal-to-epithelial transition [14]. Nevertheless, epithelial–mesenchymal transition theory is controversial and there are authors [39] who believe that this transition does not take place in cell culture.
The molecular mechanisms underlying the differentiation are not clear and it can only be speculated whether MSCs were induced to activate silent genes, or whether BTC overexpression enhanced the expression of already active genes.
It can be hypothesized that pluripotent stem cells reside in the adult bone marrow in a standby state and that these cells under certain circumstances may cross tissue (or germ-layer) boundaries to originate cells of a different lineage, challenging the notion of what it is believed—adult stem cells are committed to the germ-layer where they are derived from [15].
In our work during culture, the BTC-MSCs changed their undifferentiated status, started to produce BTC, and gained some transient elements that allow them to modify their gene expression profile. Nicotinamide—a poly(ADP-ribose) synthetase inhibitor—is also known to increase the indices of β-cells after pancreatomy [40] and it is a potent inducer of endocrine differentiation in cultured human fetal pancreatic cells [41]. Therefore, nicotinamide might play an important role in the endocrine differentiation observed in this study.
RT-PCR data showed the expression of 4 key pancreatic genes in BTC-MSCs (PDX1, NKX6.1, Pax4, and INS1) that are not expressed in MSCs.
The combined expression of PDX1 and NKX6.1 is found in the earliest pancreatic progenitor cells as well as in mature p-cells [42], indicating that BTC-MSCs have achieved expression of at least some typical β-cell genes.
The fundamental role of pancreatic p-cells is to secrete insulin in response to glucose variation. In the present study, we demonstrated the secretion of insulin from BTC-MSCs using an RIA detection technique.
Insulin secretion from BTC-MSCs was regulated by glucose; however, it occurred also in response to glucose concentrations above the physiological range. Although most of the response of normal islet occurs between 5 and 10 mM glucose, BTC-MSCs secreted most of the insulin in response to 30 mM glucose, indicating a possible difference between BTC-MSCs and p-cell, regarding the modulation of insulin release. Cell transplantation in vivo resulted in reduction of blood glucose levels, beginning 14 days posttransplantation. The glucose physiological values for rats range between 50 and 135 mg/dL [43]. In our work, diabetic animals that received bone marrow MSCs overexpressing BTC under the renal capsule presented on day 35 posttransplant reduced glycemic values compared with the control group. The amelioration observed in the animals can be attributed to the insulin production by transplanted BTC-MSCs.
In parallel, no gross tumor formation was seen in the animals as found by Fujiwaka et al. [3]. Differently, in this work, we used adult stem cells that have a lower tumorigenic ability associated with a nonviral expression system to express BTC in a safer way.
Our findings are in conflict with an early report [14] by Li et al., who could not find differentiation by the expression of BTC alone. This can be attributed to the different protocols used and also the plasticity of the MSCs. Our cells presented all the characteristics required, as stated by the International Society for Cellular Therapy [27], confirming MSC identity and plasticity. On the other hand, in agreement with our results, Hisanaga et al. have reported the in vitro differentiation of murine MSCs by the addition of BTC to the medium [44].
BTC-MSCs were capable to secrete insulin in vitro. In vivo transplanted BTC-MSCs could ameliorate the glycemic values of diabetic rats. This concept is supported by the reduced weight loss in BTC-MSC-transplanted animals.
Regarding insulin production, strategies combining BTC overexpression and other soluble factors that mimic the in vivo microenvironment could be adopted to cause the differentiated cells to produce higher levels of insulin. The pancreatic islets in the adult are among the most vascularized of all organs in the body, with a unique dense glomerular-like angioarchitecture [45]. In this way, β-cells are exposed to other cell-derived molecules that can affect the physiological regulation of glucose-induced insulin secretion. It was demonstrated that endothelial-derived molecules, like endothelin-1, thrombospondin-1, and laminins, among others, can improve β-cell function [45 –47]. These data show the importance of soluble factors secreted by islet vicinity, suggesting that the addition of soluble factors can also interfere in cell differentiation.
In agreement with Kobayashi et al. [48], we believe that, to create highly functional pancreatic β-cells such as those existing in the body, it may be necessary to reproduce the tissue structure of the pancreatic islets. Thus, the spontaneously formed 3D structures observed in our study could be a good start to improve necessary cell–cell communications.
In conclusion, our findings demonstrate the positive effects of BTC overexpression in MSCs, which can be used as a tool for cell therapy to revert hyperglycemia in a streptozotocin-induced diabetes animal model. Our results also highlight the therapeutic potential of adult bone marrow stem cells to treat diabetes.
Footnotes
Acknowledgments
This work was supported by grants from Conselho Nacional de Desenvolvimento Cientifico e Tecnólogico (CNPq), Fundo de Apoio a Pesquisa (FIPE-HCPA), Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior (CAPES), and Deutscher Akademischer Asutauschdienst (DAAD).
Author Disclosure Statement
All authors declare that there are no disclosures to be stated.