
Abstract
Because human embryonic stem (hES) cells can differentiate into virtually any cell type in the human body, these cells hold promise for regenerative medicine. The genetic manipulation of hES cells will enhance our understanding of genes involved in early development and will accelerate their potential use and application for regenerative medicine. The objective of this study was to increase the transfection efficiency of plasmid DNA into hES cells by modifying a standard reverse transfection (RT) protocol of lipofection. We hypothesized that immobilization of plasmid DNA in extracellular matrix would be a more efficient method for plasmid transfer due to the affinity of hES cells for substrates such as Matrigel and to the prolonged exposure of cells to plasmid DNA. Our results demonstrate that this modification doubled the transfection efficiency of hES cells and the generation of clonal cell lines containing a piece of foreign DNA stably inserted in their genomes compared to results obtained with standard forward transfection. In addition, treatment with dimethyl sulfoxide further increased the transfection efficiency of hES cells. In conclusion, modifications to the RT protocol of lipofection result in a significant and robust increase in the transfection efficiency of hES cells.
Introduction
H
Lipofection, among all the choices to transfer genes to cultured cells, is the simplest and least expensive technique since it does not require any specialized equipment. The standard protocol of lipofection, termed forward transfection (FT), consists of exposing cells to a DNA complex 18–24 h after seeding. Unfortunately, efficiency of lipofection in hES cells is low [6,8,11 –13]. Alternatively, in the reverse transfection (RT) protocol, the DNA complex is presented to cells just before or after seeding (Fig. 1). Here, with the objective to increase the transfection efficiency of plasmid DNA into hES cells, we introduced modifications to the RT protocol. First, we reasoned that because hES cells are anchorage-dependent cells and several of their functions are interconnected and dependent of the extracellular matrix, adding the DNA complex into the substrate would enhance their transfection efficiency. We referred to this new protocol as the modified RT (M-RT) method. This rationale is supported by previous findings showing that adenovirus contained within hydrogels [21] or immobilized in biomaterial surfaces [22,23] enhance transduction of fibroblasts. Because cell endocytosis plays a major role in plasmid DNA incorporation by lipofection, we also tested if low concentrations of dimethyl sulfoxide (DMSO) would increase the transfection efficiency of hES cells.
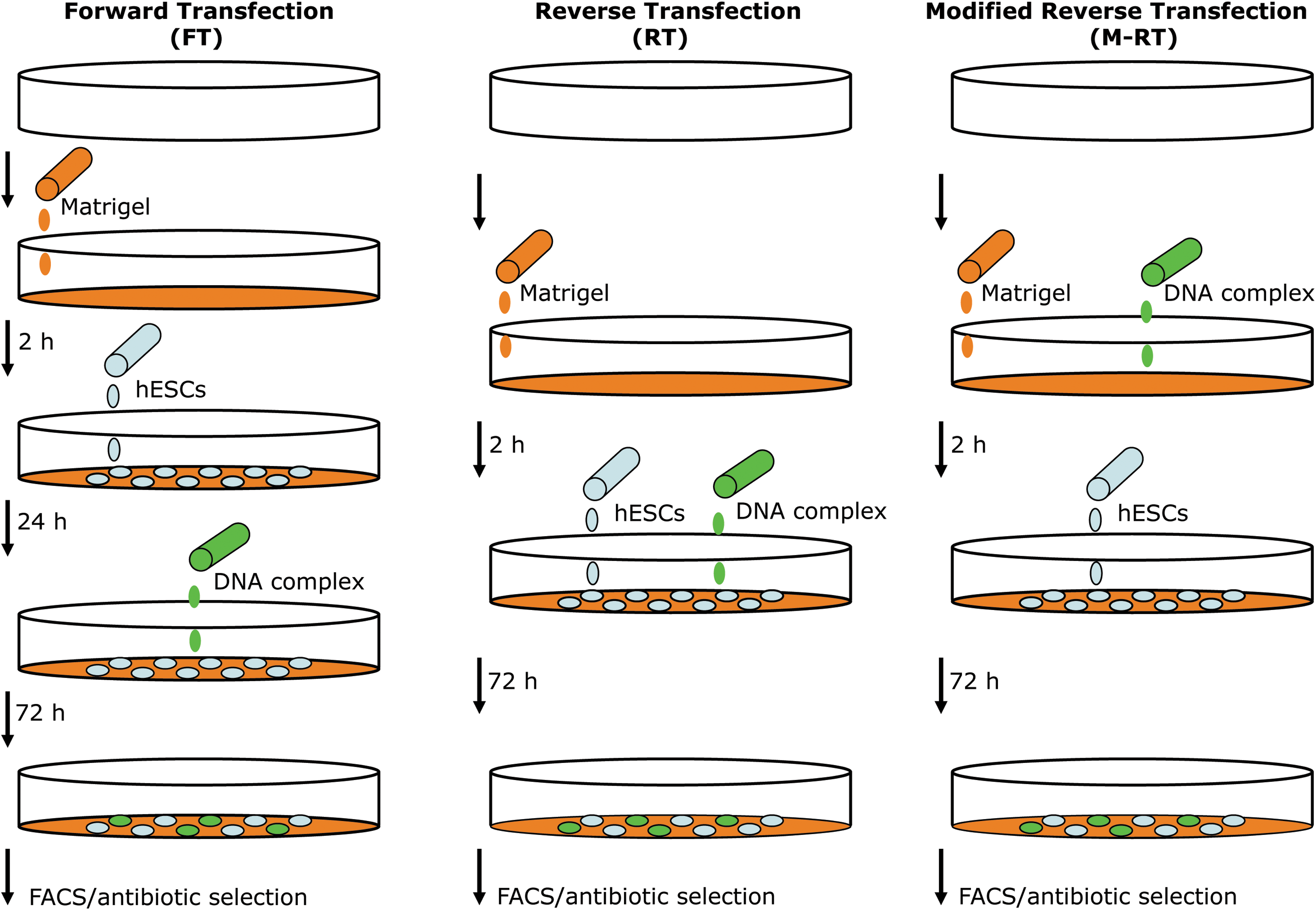
Schematic representation of the forward transfection (FT), reverse transfection (RT), and modified RT (M-RT) gene transfer protocols. In the FT protocol, cells are seeded on Matrigel-coated tissue culture plates and transfection is performed 18–24 h after the addition of the DNA complex to the culture medium. In the RT protocol, the DNA complex is added to the culture medium at the same time that cells are seeded. However, in the M-RT protocol, the DNA complex is added to Matrigel solution during the 2-h incubation time required to coat tissue culture plates with Matrigel. In the scheme, a white three-dimensional circle represents a tissue culture plate, while Matrigel solution and gel coating is shown in orange. The DNA–transfection mix is shown in green, and human embryonic stem cells (hESCs) are depicted as blue ovals. In addition, the time required between steps in each protocol is indicated in hours (h). FACS, fluorescence activated cell sorting.
Transfections were performed on hES cells with several reporter plasmids of different molecular weights, and flow cytometry was used to calculate the transfection efficiency of each transfection protocol. The determination of transfection efficiency consisted of 2 parameters: the percentage of transfected cells and the mean fluorescence intensity (MFI) of the protein produced by the reporter gene in transfected cells. In addition, clonal stable cell lines were established by transfection of hES cells with a plasmid encoding enhanced green fluorescent protein (EGFP) and a neomycin (NEO) selectable marker. Our results demonstrate that transfection efficiency was doubled in both transient and stable transfections of hES cells using the M-RT protocol compared to FT and RT. In addition, treatment with DMSO further increased the percentage of reporter expressing cells.
Materials and Methods
Cell culture
hES cell lines H9:WA09 and BG01 were grown in coculture with mitotically inactivated mouse embryonic fibroblasts (MEFs) from CF-1 mice on gelatin-coated plates. MEFs were used at a density of 25,000 cells/cm2. The culture medium for hES cells consisted of Dulbecco's modified Eagle's medium (DMEM)/F12 (Invitrogen) supplemented with 4 ng/mL β-fibroblast growth factor (FGF; Invitrogen), 20% knock-out (KO) serum replacement (Invitrogen), 1 mM
Transfection assays were performed on cells cultured on Matrigel-coated plates and with MEF-conditioned medium (MEF-CM) supplemented with β-mercaptoethanol (0.1 mM),
Plasmid DNA
Plasmid constructs ks-dsRED, pEGFP-N1, pu52-EGFP, and pCEP-EGFP were used for transfections assays. Plasmid DNA was purified on Qiagen midi prep columns (Qiagen) and sterilized using a 0.22 μM filter. Superhelicity of DNA for transfection experiments was determined by electrophoresis on 0.7% agarose–ethidium bromide gels. Only highly supercoiled (>90%) preparations of DNA were used for transfection.
ks-dsRED is a 4.5-kb plasmid that contains the coding region of the dsRED protein (Clontech) under the transcriptional control of a minimal cytomegalovirus (CMV) promoter (from pCEP). It was cloned in a modified pBSKS-II (Stratagene) vector that contains the SV40 late polyadenylation signal downstream of the dsRED gene.
pEGFP-N1 is a 4.4-kb plasmid (Clontech) that contains the coding sequence of the EGFP under the control of a minimal CMV promoter and an SV40 late polyadenylation signal. The plasmid also contains a dual prokaryotic/eukaryotic kanamycin/NEO selectable marker.
pu52-EGFP is a 4.9-kb plasmid that contains the coding sequence of EGFP under the transcriptional control of the human UBC promoter (a 1.2-kb fragment of the UBC gene, nucleotides 123964272–123965484 from human chromosome 12) and a NEO selectable marker, in pBSKS-II. This plasmid was a generous gift from Dr. Sue O'Shea in the Cell and Developmental Biology Department at the University of Michigan Medical School.
pCEP-EGFP is a 11-kb plasmid that contains the coding sequence of the human EGFP cloned in vector pCEP4 (Invitrogen). Thus, EGFP is expressed under the control of a CMV promoter and an SV40 late polyadenylation signal.
Lipid-based transfection assays
Fugene6 (Roche), Lipofectamine (Invitrogen), and ExGen500 (Fermentas) were used following instructions provided by each manufacturer. The FT assay was performed as described previously [24] except that hES cells were harvested using TrypLE Select (Invitrogen) and treated with the ROCK inhibitor Y-27632 (10 μM; Alexis Biochemicals) 1 h before harvesting and during the first 24 h after plating, to increase cell survival after dissociation [7]. Briefly, hES cells were collected with TrypLE Select, counted, and seeded at dilutions ranging from 20,000 to 60,000 cells/cm2 on Matrigel-coated plates. Sixteen to 18 h postseeding the DNA complex was added to transfect cells. The DNA complex per well of a 6-well tissue culture plate consisted of 2 μg of plasmid DNA, 97 μL of Optimem (Invitrogen), and the required amount of each of the transfection reagents. Twenty-four hours posttransfection, the medium was replaced.
The RT was conducted as described above and the DNA complex was added simultaneously as hES cells were seeded. The M-RT was conducted as described above; however, the DNA complex was added to the Matrigel solution 0, 30, 60, and 90 min after the initiation of the 2-h incubation for tissue culture plate coating. We optimized the protocol using Fugene6 (6 μL per well in a 6-well tissue culture plate). In some experiments, DMSO (Sigma) was used at 1% or 2% (v/v) in the culture medium during the first 24 h posttransfection.
Nucleofection transfection assays
Nucleofection of hES cells was performed as described previously [24]. As above, hES cells were harvested using TrypLE Select and treated with the ROCK inhibitor Y-27632 1 h before harvesting and during the first 24 h after plating [7]. Briefly, we used the V-Kit solution (Amaxa) and applied program number A-23 in the nucleofector device (Lonza). We routinely transfected 4 × 10 6 cells using 4 μg of each plasmid DNA.
Flow cytometry analysis
Seventy-two hours posttransfection, cells were harvested using trypsin 0.5% (Invitrogen), and analyzed by flow cytometry to determine the percentage of reporter expressing cells, and the MFI of the FP signal. Analysis was carried out with FACSscan (Becton Dickinson) using standard procedures. Background fluorescence and autofluorescence were determined using nontransfected cells as control. The number of FP-positive cells and the arithmetic mean channel number as a measure of the MFI within the same population were normalized to nontransfected cells. At least 3 independent experiments were performed in duplicate for each experiment.
Establishment of stable transfected hES cell lines
To generate clonal cell lines, hES cells were transfected with either the linearized pEGFP-N1 plasmid (using Afl II) or the pu52-EGFP plasmid. Four days after transfection hES cells were selected with geneticin (G418; Invitrogen) for 2 weeks. Transfected hES cells were treated with 50 and 100 μg/mL of G418 during the first and second week, respectively, as previously described [9]. G418-resistant hES cell-foci were manually dissected using a pulled Pasteur pipette, and transferred to a well of a 12-well tissue culture plate containing irradiated MEFs and cultured in hES cell media. Fully developed and undifferentiated hES colonies were subsequently manually passaged, and frozen as previously described [24].
Immunocytochemistry analysis
Cells were fixed for 30 min at room temperature using 2% paraformaldehyde in phosphate-buffered saline (PBS). Subsequently, cells were permeabilized with a 0.2% Triton X-100 (Sigma) solution in PBS for 10 min, blocked with 1% donkey serum (Jackson ImmunoResearch) in PBS containing 0.1% Triton X-100 for 30 min in a humidified chamber, and incubated overnight with the primary antibody in PBS/0.1% Triton X-100. On the next day, plates were washed twice with PBS/0.1% Triton X-100, and incubated with a secondary antibody (in PBS/0.1% Triton X-100) for 30 min in a humidified chamber. Plates were washed twice with PBS/0.1% Triton X-100, and once with PBS. The following antibodies were used at the indicated dilutions: Oct4 (1/200; Santa Cruz Biotechnology), Sox-2 (1/200; Millipore), Tra-1-60 (1/50; Santa Cruz Biotechnology), and SSEA4 (1/100; Santa Cruz Biotechnology). Cy3-conjugated anti-goat and anti-mouse secondary antibodies (Jackson ImmunoResearch) were used at a 1/200 dilution, and DAPI (1/10,000; Sigma) was used to stain nuclear DNA.
In vitro hES cell differentiation assay
The pluripotency of stably transfected hES clones was evaluated using a standard embryoid body (EB) formation assay [24]. Briefly, undifferentiated hES cells were cultured in suspension with the hES cell culture medium lacking β-FGF for 10 days to generate mature EBs. Then, EBs were harvested and total RNA was isolated.
RNA extraction and purification
Cells were collected by centrifugation at 800 g and then disrupted by vigorous pipetting in 1 mL of Trizol Reagent (Invitrogen). Chloroform (200 μL) was added to this solution followed by centrifugation (∼13,000 g). The aqueous phase containing RNA was removed and additionally purified using the RNeasy Mini-Kit (Qiagen) following the manufacturer's RNA Clean-up protocol with the optional On-column DNase treatment. RNA quality was determined using RNA 6000 Nano Assays performed on the Bioanalyzer 2100 Lab-on-a-Chip system (Agilent Technologies).
Reverse-transcription polymerase chain reaction analysis
Total RNA was reverse transcribed using SuperScript™ One-Step reverse transcriptase (RT)-polymerase chain reaction (PCR) with platinum® Taq (Invitrogen). In a single reaction (50 μL), 1 μg of total RNA and 20 pmol of forward (F) and reverse (R) primers were used. The cDNA synthesis and predenaturation were carried out in the first cycle at 48°C for 45 min, followed by a second cycle at 94°C for 2 min. PCR amplifications were performed for 40 cycles at 94°C for 15 s, 55°C for 30 s, and 72°C for 30 s. An initial denaturation step (94°C for 2 min) and a final extension cycle (72°C for 10 min) were also included. Finally, 10 μL of each PCR was loaded onto a 1.0% agarose gel and size-fractionated. The following genes were analyzed: β-actin (F, 5′ atctggcaccacaccttctacaatgagctgcg 3′; R, 5′ cgtcatactcctgcttgctgatccacatctgc 3′), BMP4 (F, 5′ tgagcctttccagcaagttt 3′; R, 5′ cttccccgtctcaggtatca 3′), Nestin (F, 5′ cagctggcgcacctcaagatg 3′; R, 5′ agggaagttgggctcaggactgg 3′), and AFP (F, 5′ ccatgtacatgagcactgttg 3′; R, 5′ ctccaataactcctggtatcc 3′).
Cytogenetic analysis
Karyotype analysis of stably transfected hES cell lines was performed at Cell Line Genetics. Chromosomes were prepared using standard protocols and measurements were performed using the Giemsa/Trypsin/Leishman (GTL)-banding method on at least 20 metaphase preparations.
Statistics
All experiments were performed at least in triplicate. Mean ± standard deviation values were calculated and analyzed by Student's t-test, setting the significant value at 0.05.
Results
As an initial attempt to identify the most efficient method to transfect hES cells, we compared the nucleofection technology to FT lipid-based methods (Fugene6, Lipofectamine, and ExGen500). Reporter plasmids that expressed EGFP in transfected cells and that could be quantified by flow cytometry analysis were used to evaluate and compare the transfection efficiency. As plasmid size has been related to transfection efficiency [24], 2 reporter plasmids that differed in size (4.4 and 11 kb) were used. The transfection efficiency was analyzed by flow cytometry 72 h posttransfection. As expected, the transfection efficiency was inversely correlated to the size of the reporter in all the methods used (Supplementary Fig. S1, available online at
The M-RT protocol is an efficient method to transfect hES cells
Aiming to increase transfection efficiency of hES cells with lipid base technology, we introduced modifications to the RT protocol and analyzed if the combination of DNA transfection mixed with Matrigel before hESC seeding would increase the exposure of cells to plasmid DNA and enhance its subsequent uptake (Fig. 1). All experiments were performed using the hES cell lines H9 and BG01, and because results among cell lines were similar, they were pooled for analysis. Similarly, reporter plasmids that expressed FPs in transfected cells and that could be quantified by flow cytometry analysis were used to compare the transfection efficiency of hES cells using M-RT to the standard FT and RT protocols. In the M-RT method, the plasmid DNA complex was added to Matrigel solution 0, 30, 60, and 90 min after the initiation of the 2 h incubation for tissue culture plate coating (Fig. 1). Undifferentiated hES cells were transfected with a 4.2-kb plasmid expressing a red FP (dsRED). Compared to FT and RT methods, the M-RT method demonstrated superior and significant (P ≤ 0.05) transfection efficiency of hES cells (Fig. 2). Using FT and RT, 7.1% and 11.9% of cells were identified as dsRED positive (+), respectively. In contrast, DNA complexes embedded within Matrigel in the M-RT transfection method increased the percent of dsRED (+) cells to 19.6%, 15.31%, 12.7%, and 12.1% depending on the time at which the DNA complex was added to Matrigel (0, 30, 60, or 90 min, respectively) after the gel-coating process was initiated. A significant (P ≤ 0.05) increase in MFI was also detected in hES cells transfected with the M-RT method (Fig. 2). Thus, the M-RT protocol reproducibly increased the transfection efficiency of hES cells.
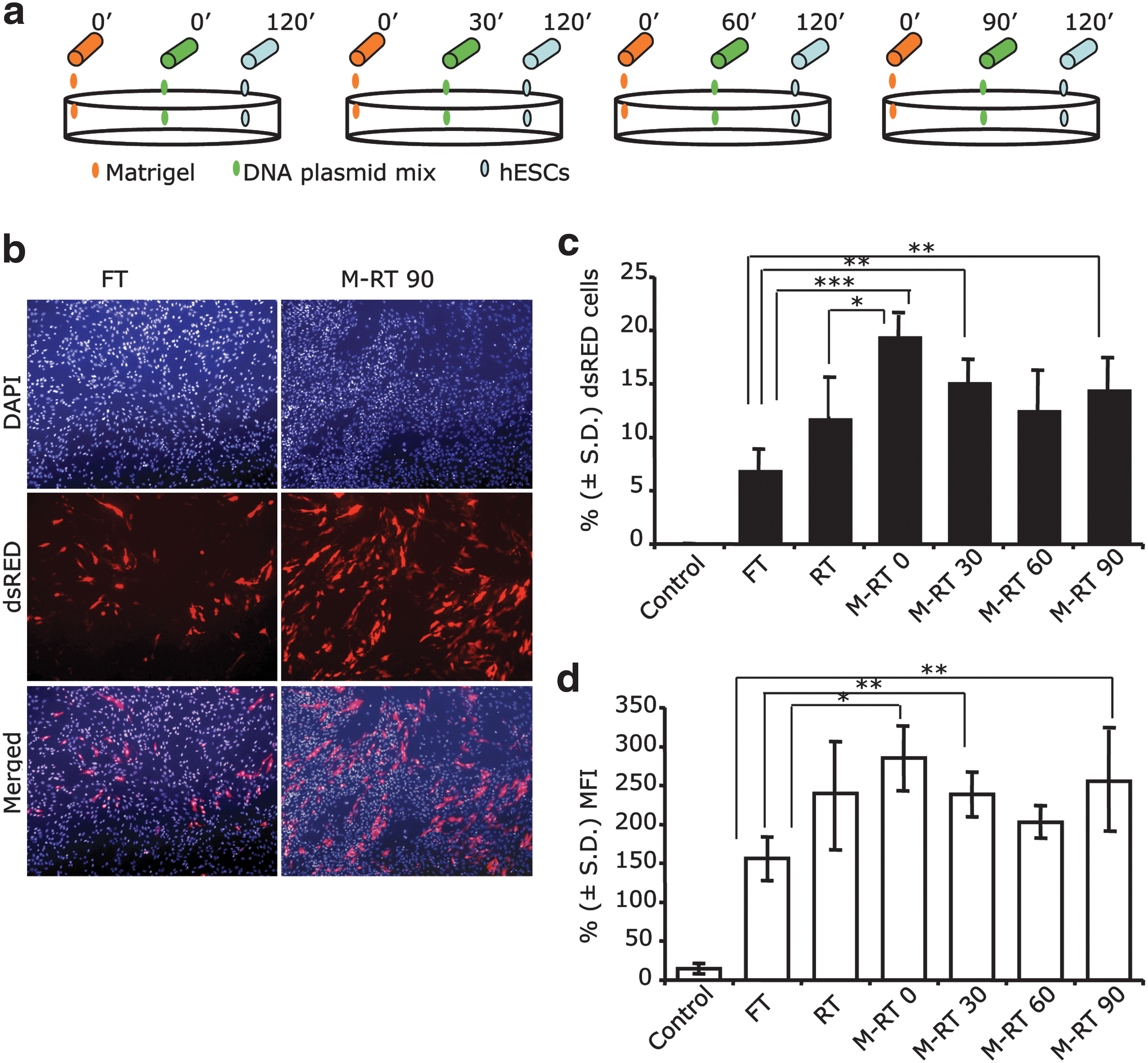
Modified reverse transfection (M-RT) enhance the transfection efficiency of human embryonic stem cells (hESCs). (
The M-RT protocol is an efficient method to transfect hES cells with high-molecular-weight DNA constructs
Because hES cells are notoriously difficult to transfect and results vary depending on the size of the construct used [6,8,11 –15], we next determined if M-RT would be useful to transfect DNA constructs of larger size. Thus, we tested the transfection efficiency of hES cells obtained by M-RT and FT using an 11-kb plasmid that contained a EGFP reporter (pCEP-EGFP; see Materials and Methods section). We found that the transfection efficiency of the 11-kb plasmid into hES cells was significantly (P ≤ 0.05) enhanced when using M-RT (13.14%) compared to FT (7.5%; Fig. 3). Thus, the M-RT method is useful to transfect hES cells using plasmids of different sizes.

Modified reverse transfection (M-RT) using a 11-kb reporter plasmid. The graph shows the percentage ± standard deviation (S.D.) (n = 3) of enhanced green fluorescent protein (EGFP) expressing cells transfected with plasmid pCEP-EGFP (11-kb; see Materials and Methods section) using forward transfection (FT) or M-RT at 90 min (indicated in the graph). Nontransfected cells were used as a control group (UTF), and flow cytometry was used for quantification. UTF, untransfected.
DMSO treatment increases the percentage of transfected hES cells
Next, we tested whether cell membrane permeabilization using low concentrations of DMSO would further increase the transfection efficiency of hES cells. We used the dsRED-expressing plasmid and the M-RT protocol with DNA complex addition 90 min after the initiation of Matrigel coating. Treatment with 1% and 2% DMSO during the first 24 h post-hES cell seeding resulted in a significant (P ≤ 0.05) increase in the percent of dsRED (+) cells (18.96% and 20.36%, respectively) compared to no DMSO treatment (12.19%; Fig. 4). However, no increase in the mean fluorescent signal intensity of transfected cells was observed upon DMSO treatment. DMSO at 2% was mildly toxic to hES cells, as fewer surviving cells were observed 72 h posttransfection (data not shown).

Dimethyl sulfoxide (DMSO) treatment increases the transfection efficiency of human embryonic stem cells (hESCs) achieved by the modified reverse transfection (M-RT) protocol. (
Generation of stable clonal hES cell lines using the M-RT method
Genetic modification of hES cells usually requires the stable integration of transfected DNA into the cell genome. Therefore, we compared the integration efficiency of plasmid DNA in hES cells using the FT and M-RT protocols. A linearized plasmid (EGFP-N1) that contains the EGFP gene and a NEO resistance cassette was used. Transfected cells were treated with G418 for 2 weeks to select clonal cell lines with stably integrated plasmid DNA. Consistent with the transient transfection results, a 2-fold increase in the number of G418-resistant colonies was observed with the M-RT method compared to the FT method (Fig. 5). Similarly, using a supercoiled construct (pu52EGFP; see Materials and Methods section), an ∼2-fold increase in the number of stable clonal cell lines was observed when hES cells were transfected using M-RT compared to FT (data not shown).

The modified reverse transfection (M-RT) protocol doubles the establishment of clonal human embryonic stem cell (hESC) lines. hESCs were transfected using either the forward transfection (FT) protocol or the M-RT 90 protocol with a plasmid that contains a dual prokaryotic/eukaryotic kanamycin/neomycin selectable marker cassette (pEGFP-N1). Seventy-two hours posttransfection, treatment with G418 was initiated to select G418-resistant hESC foci. (
To confirm that genetically modified clonal hES cell lines obtained by M-RT methods truly behave as hES cells, we expanded a clonal cell line containing the plasmid pu52EGFP in its genome (H9-52-EGFP). Characterization of H9-52-EGFP cells at passage 5 demonstrated high expression levels of EGFP and identical characteristics to the parental H9-hES cell line, such as expression of hES cell markers Oct3/4, Sox-2, Tra-1-60, and SSEA-4 (Fig. 6a). The H9-52-EGFP cells had a normal female karyotype with 46 chromosomes examined at metaphase (Fig. 6b). To determine the differentiation potential of H9-52-EGFP cells, we conducted an in vitro EB formation assay and observed that H9-52-EGFP cells generated EBs at a similar rate to the parental cell line and also expressed high levels of EGFP (Fig. 6c). RT-PCR analysis confirmed expression of ectoderm, endoderm, and mesoderm markers in the isolated RNA fraction of EBs generated from H9-52-EGFP cells (Fig. 6d).
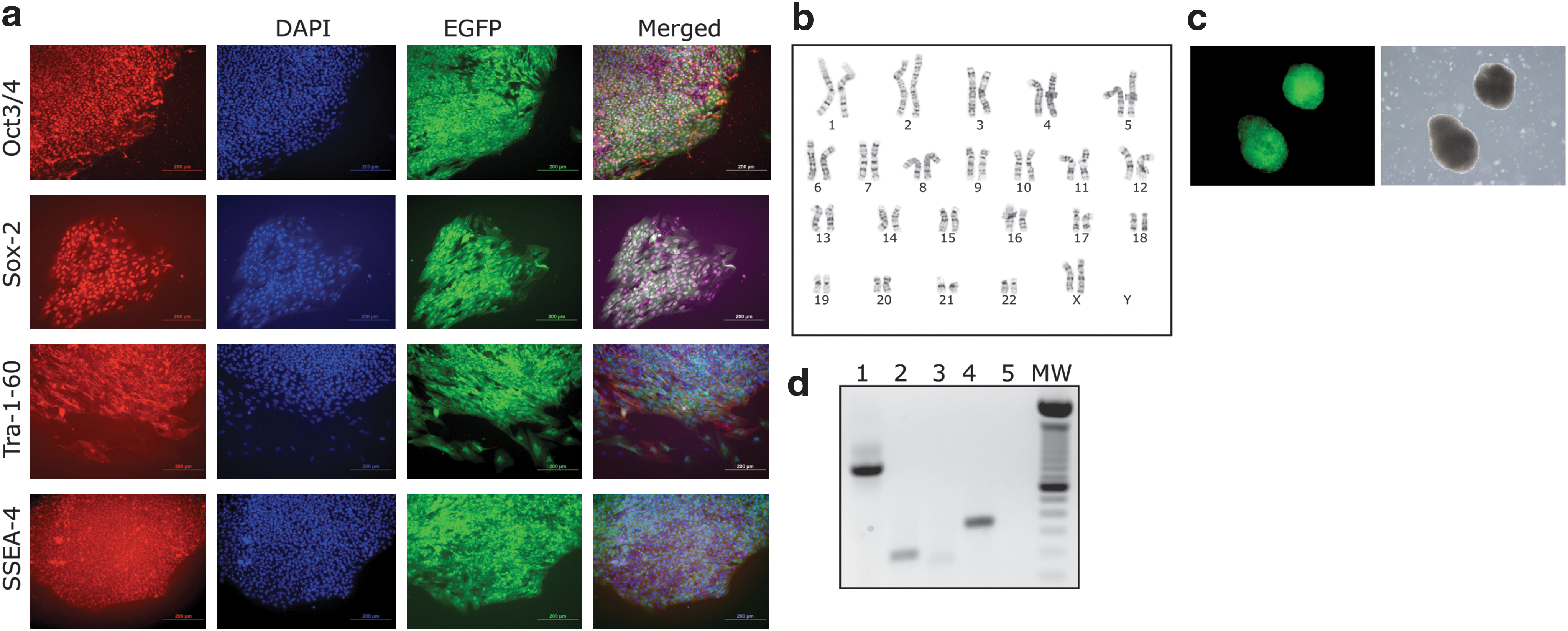
The modified reverse transfection (M-RT) protocol generates clonal human embryonic stem cell (hESC) lines. (
Discussion
If hES cells are to reach their presumed potential for regenerative medicine, effective and predictable protocols to grow, differentiate, and manipulate their genome must be developed. Several of the standard gene transfer protocols have been adapted for hES cells and the results have been variable depending on the method used. Among them, nucleofection is generally the most effective method (Supplemental Fig. S1). Unfortunately, nucleofection protocols require access to a specific nucleofection device, and frequently results in a high percentage of cell death. In contrast, the toxicity and variability of lipid-based methods are factors that also negatively affect the success of hES cell transfection [6,8 –10,25]. Additional strategies like the use of viruses have also been developed for hES cell transgenesis [17,18,20,26], but the efficiency of gene transfer to hES cells is much lower than in differentiated cells.
Here, we developed a simple and efficient new protocol based on lipid-mediated transfection technology to transfect hES cells using Fugene6; however, other lipid-based reagents likely could be adapted to the M-RT protocol described here. The standard RT protocol of lipofection was modified to achieve at least a doubling in the transfection efficiency of hES cells compared to standard protocols, and similar to the obtained with nucleofection technology. One modification consisted in incorporating the DNA complex in Matrigel, a substrate on which hES cells thrive in a feeder-free environment [27]. The absence of feeder cells in this culture condition has the benefit of less cellular competition for transfection reagents. Taking advantage of the need to coat tissue culture plates with Matrigel, DNA complexes were suspended in the Matrigel solution at different time points before cell seeding. Thus, in the M-RT method, the Matrigel substrate carried the DNA complexes, whereas in the standard FT and RT methods, the DNA complexes are delivered to attached cells or cells in suspension, respectively.
Maximum transfection efficiency was observed when the DNA complex was mixed in the Matrigel solution at the beginning of the 2 h matrix-coating process. The transfection efficiency gradually decreased in groups where the DNA complex was added 30, 60, and 90 min after Matrigel coating was initiated. However, the transfection efficiency was still higher than that obtained with FT and RT methods. Previously, it has been demonstrated that serum components and incubation in the liquid medium at 37°C negatively influence adenoviral viability [21,28]. Thus, it is possible that DNA complexes contained within Matrigel are protected from medium components, contributing to the increased transfection rates.
Our results also suggest that transient permeabilization of cell membranes with DMSO significantly improves transfection efficiency of hES cells. However, the concentration of DMSO is critical, as we observed increased cell death at 2% DMSO, while transfection efficiency with 1% DMSO was augmented without sacrificing cell viability. The beneficial effect of DMSO in transfecting mammalian cells has been described previously using electroporation [29,30], but not with lipofection or when using hES cells. It has been suggested that cellular membranes are more permeable and stable under the influence of DMSO, thus ensuring a better transfection and survival rate of cells after electroporation. During the transition from interphase to mitosis, cell membrane tension increases dramatically, preventing the invagination of endocytic vesicles and reducing endocytosis [31]. Conversely, amphyphilic compounds such as DMSO, deoxycholate, and ethanol reduce membrane tension and enhance endocytosis. As the cell population doubling of hES cells is ∼36 h [32], it is likely that the majority of cells are in the transition from interphase to mitosis during the 24 h transfection period. Thus, it is possible that the increased transfection rate observed in hES cells treated with DMSO is due to increased cell endocytosis and uptake of DNA complexes contained within the Matrigel substrate. Although it is possible that hES cell differentiation could be induced with DMSO, we did not observed this. When 1% DMSO was used, stably transfected and pluripotent hES cell lines were generated (Fig. 6).
Transfection of 2 different hES cell lines with both linearized and supercoiled constructs of various sizes indicated that M-RT is a simple and reproducible method to genetically manipulate hES cells. In addition, the efficiency of the M-RT method was also demonstrated by doubling the number of stable clonal colonies obtained after antibiotic selection. The stable cell lines generated using the M-RT method behaved as true hES cells, as they continued to proliferate in an undifferentiated state in culture while expressing the inserted reporters genes (EGFP and NEO), expressed markers of pluripotency, displayed a stable karyotype, and maintained their capacity to differentiate into the 3 germ layers.
Conclusions
Modifications to a standard transfection protocol of lipofection, such as incorporating the DNA complex into the substrate as well as DMSO treatment, lead to robust transfection rates of hES cells, similar to obtained with nucleofection technology. The M-RT protocol of lipofection will be useful in research aimed to genetically manipulate hES cells.
Footnotes
Acknowledgments
We thank the University of Michigan Stem Cell Center for tissue culture support, and Dr. Theresa Grastch and Dr. Sue O'Shea for generous gift of plasmid pu52-EGFP. Research in J.L.G.-P. laboratory is supported by ISCIII-CSJA (EMER07/056), a Marie Curie IRG action (FP7-PEOPLE-2007-4-3-IRG), and a Miguel Servet Contract (CP07/00065,ISCIII), as well as by CICE (P09-CTS-4980) and Proyectos de Investigación en Salud PI-002 both from the Junta de Andalucia, Spain, and throughout the Spanish Ministry of Health (FIS PI08171). Research in P.H.K. laboratory is supported by the National Institutes of Health (R01 DE016530).
Author Disclosure Statement
The authors declare that no competing interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.